Full HTML
Cardiorenal, Renocardiac, and Reno-Cardio-Cardiac Syndromes: An Updated Review on General Definitions, Pathophysiology, and Therapies (Part 1)
Elmukhtar Habas1, Ala Habas2, Amnna Rayani3, Aml Habas4, Gamal Alfitori5, Eshrak Habas2, Almehdi Errayes5, Kalifa Farfar6, Anand Kartha5, Abdel-Naser Elzouki5
Author Affiliation
1 Senior Consultant, Department of Medicine, Hamad General Hospital, Prof of Internal Medicine, Qatar University, Doha-Qatar, Open Libyan University, Libya
2 Resident, Department of Medicine, Tripoli Central Hospital, University of Tripoli, Tripoli-Libya,
3 Professor of Pediatric Medicine, Senior Consultant, Tripoli Children Hospital, Open Libyan University, Tripoli-Libya,
4 Resident, Tripoli Children Hospital, Open Libyan University, Tripoli-Libya,
5 Senior Consultant, Department of Medicine, Hamad General Hospital, Doha, Qatar,
6 Consultant, Department of Medicine, Alwakra General Hospital, Qatar.
Abstract
Background: Acute and chronic heart or kidney failure affect each other in cardiorenal syndromes (CRS). In CRS, hemodynamic and non-hemodynamic changes occur, causing acute or progressive renal and cardiac failures. CRS is classified into five types based on the first organ failure and causes failure of the other organ. We believe that the current CRS classification is not the correct one that effectively describes the underlying cause of CRS. Hence, we consider it better to be classified into three categories (cardiorenal, renocardiac, and cardio-reno-cardiac syndrome) and then subdivided into acute and chronic types or types 1 and 2 (respectively, according to the onset of the underlying type of failure (i.e., acute or chronic). Other subtypes that occur in the heart and dysfunction occur simultaneously are acute cardio-reno-cardiac syndrome (type 5) and Chronic cardio-reno-cardiac syndrome (type 6).
Aim: In Part 1 of the review series, the pathophysiological mechanisms and clinical and therapeutic applications of all types of CRS will be narratively discussed and updated. Furthermore, we provide a comprehensive review of diagnostic biomarkers and their clinical significance in the identification, outcome prediction, and treatment of all CRS types.
Method: An extensive search of PubMed, Google, EMBASE, Scopus, and Google Scholar was conducted for review articles, original articles, and commentaries published between Jan 2010 and Aug 2024 using different phrases, texts, and keywords, such as CRS, renocardiac syndrome, and CRS. The topics included secondary CRS, CRS pathogenesis, CRS therapy, SLGT inhibitor use in CRS, novel therapy in CRS types, and prevention of CRSs.
Conclusion: Renal and cardiac failure in patients with CRS seem to have different pathophysiological mechanisms. Early detection and treatment can improve the outcomes of CRS. Clinical manifestations and therapy protocols vary according to pathophysiology. Hence, new guidelines and research on universal diagnostic and treatment techniques are urgently required. Moreover, the current nomenclature for CRS is confusing; therefore, we believe that a new nomenclature system should be introduced, reducing confusion and making differentiation between CRS types easier and less confusing.
DOI: 10.63475/yjm.v4i1.0028
Keywords: Chronic kidney disease, acute decompensated heart failure, Cardiorenal syndrome, Renocardiac syndrome
Pages: 9-42
View: 48
Download: 70
DOI URL: https://doi.org/10.63475/yjm.v4i1.0028
Publish Date: 21-05-2025
Full Text
The term "heart and kidney interaction" or cardiorenal syndrome (CRS) emerged in 2004, and subsequent research has consistently shown an increased prevalence of cardiac and renal disorders. Research has shown that cardiorenal and renocardiac (CRS, RCS, respectively) often occur and have a substantial impact on the rate of death, morbidity, intricacy, and healthcare expenses.[1–3]
Kidneys and the heart have a reciprocal association, meaning that both organs experience similar physiological and pathological situations. The heart depends on the kidney-controlled balance of the body's internal environment, which includes electrolytes, fluids, and toxins. In contrast, the kidney's function significantly depends on regulating blood flow and volume via neurohormonal, hemodynamic, and inflammatory processes.[4]
CRS is a recognized pathological disorder that affects both the kidneys and the heart. CRS is a medical disorder that mostly arises in the case of sudden or severe malfunction in one organ, resulting in severe or acute malfunction of the other organ. In 2004, the National Heart, Lung, and Blood Institute Working Group conducted a comprehensive assessment to examine the relationship between the kidneys and heart. Based on this assessment, CRS is described as the outcome of kidneys and other circulatory compartment interaction that leads to an intravascular volume increase, exacerbating heart and kidney damage, producing kidney and heart failure (HF).[5,6] CRS is a term used to describe the strong connection between cardiovascular (CV) and renal disorders and the probabilities of their reciprocal effects in causing their progression.[6–8] CRS is divided into 5 types, of which types 1 and 2 represent acute and chronic HF, leading to acute or chronic renal failure, respectively. [6,9]
CRS was nominated in 2004 by the Working Group of the National Heart, Lung and Blood Institute. Initially, they divided CRSs into four types (type1-4). Type 1 and type 2 CRS are known to be acute (aCRS) and chronic (cCRS). aCRS is defined as acute kidney injury (AKI) resulting from acute heart dysfunction. The cCRS is a condition that is characterized by a slow progression of renal dysfunction due to the progression of heart dysfunction.[10,11]
Later, it was observed that some individuals with acute or chronic kidney dysfunction developed acute or heart dysfunction. They are referred to as type 3 and 4 CRS, respectively. Both types had kidney dysfunction that led to cardiac dysfunction. This condition is known as renocardial syndrome (RCS). Furthermore, RCS was classified into type 3 (acute) and type 4 (chronic) CRS. In Type 3 (acute), AKI leads to acute dysfunction in a previously healthy heart. Whereas, in chronic (type 3) CRS, due to progressive chronic kidney disease (CKD), heart function progressively deteriorates. Although types 3 and 4 CRS are misnomers and confusing, we think it is better to call them acute RCS (aRCS) and chronic RCS (cRCS), respectively.
Additionally, it has been noted that systemic chronic diseases damage both the heart and kidneys simultaneously, causing what is known as type 5 CRS (secondary CRS).[10,11] According to the chronicity of the underlying systemic process and timing of renal or cardiac dysfunction, CRS-5 is further classified as acute or chronic. Recently, acute secondary CRS has been renamed as type 5 CRS, and chronic type 5 was known as type 6 CRS.[12] Again, we find that this is also confusing; hence, we suggest naming this type as a cardio-reno-cardiac syndrome (CRCS) and can be subdivided as others into acute (aCRCS) and chronic CRCS (cCRCS). Figure 1 shows the nomenclature and classification of CRS.
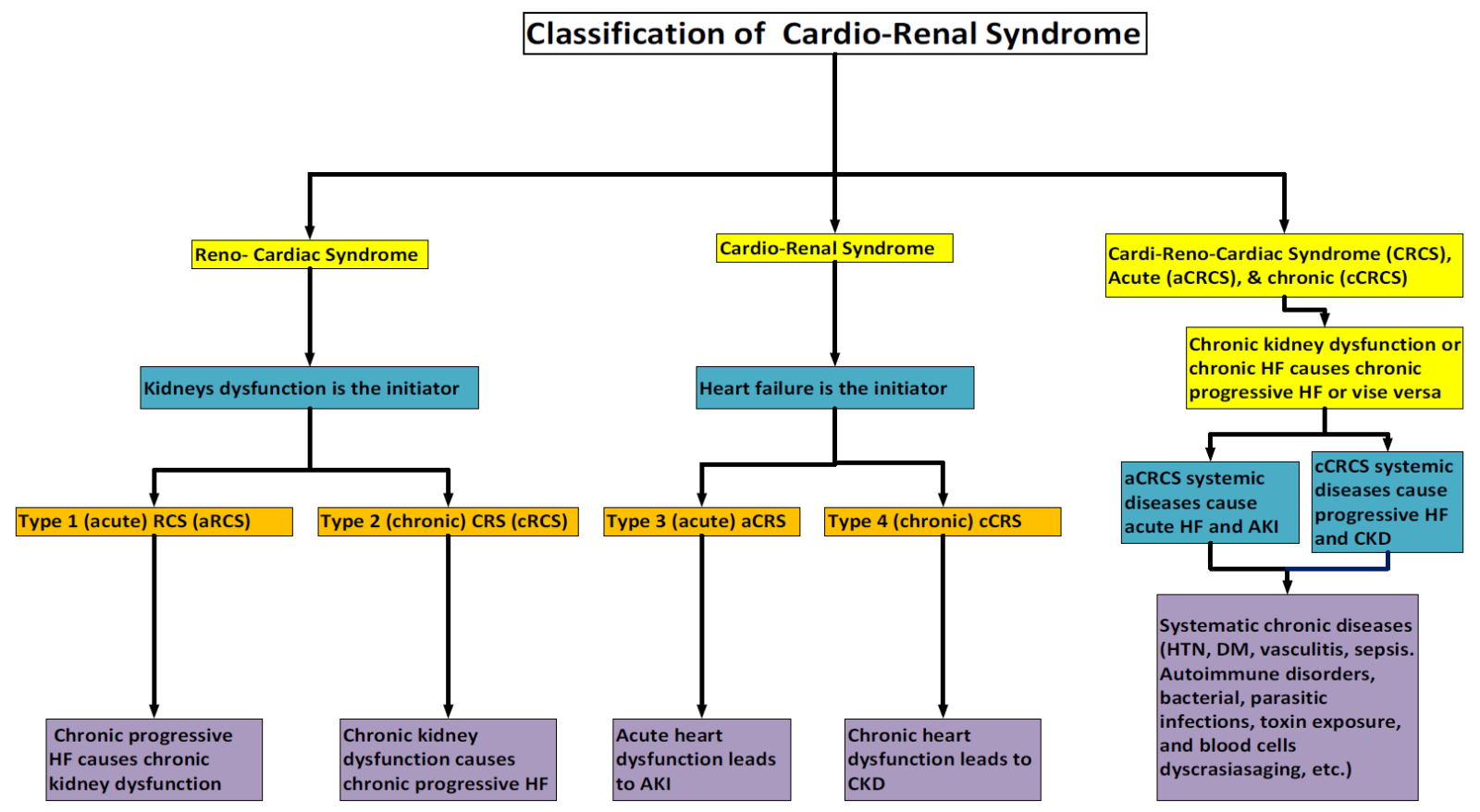
Figure 1. Nomenclature and the classification of CRS.
Abbreviation: Cardiorenal (CRS), renocardiac (RCS), chronic kidney disease (CKD), heart failure (HF), and acute kidney injury (AKI).
There has always been a conflict in AKI definition until recently, in which acute renal impairment was unambiguously defined utilizing the Acute Kidney Injury Network (AKIN) and risk, injury, failure, loss of kidney function, and end-stage kidney disease (RIFLE) classifications.[13] Whereas Acute heart dysfunction refers to two separate disorders: cardiogenic shock and acute heart failure (AHF). These two conditions differ in clinical and pathophysiological aspects.[9] Table 1 provides a detailed list of these phenotypes, including the new nomenclature, and a brief description of the underlying cause.
Table 1. CRS and RCS Classification
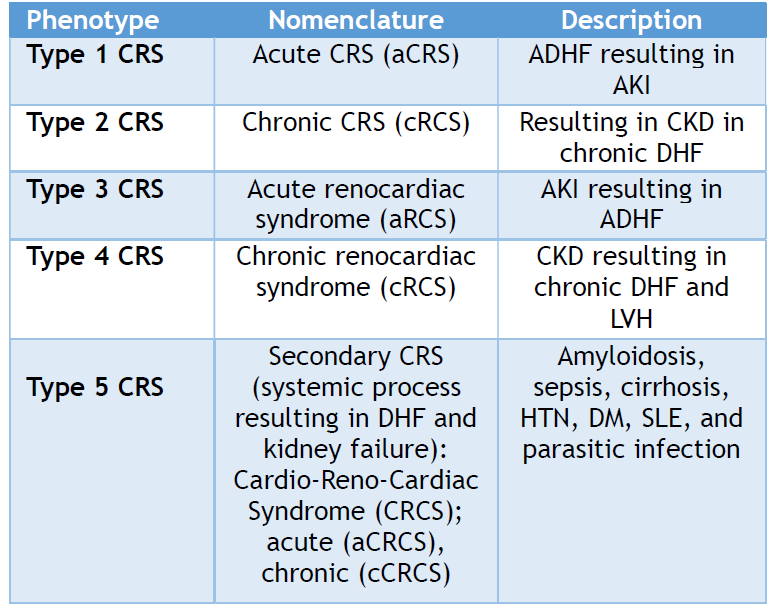
Abbreviations: Cardiorenal syndrome (CRS), chronic kidney disease (CKD), heart failure (HF), acute kidney injury (AKI), chronic decompensated (DHF), hypertension (HTN), diabetes mellitus (DM), systemic lupus erythematosus (SLE), chronic kidney disease (CKD), left ventricular hypertrophy (LVH), acute decompensated heart failure (ADHF), and left ventricular hypertrophy.
Furthermore, another classification scheme was proposed for CRS. The scheme categorized CRS based on different clinical manifestations, regardless of the initial organ affected. This classification includes manifestations such as hemodynamic compromise, uremic or vascular symptoms, neurohumoral disturbances, anemia, bone mineral metabolism disruptions, and the malnutrition inflammation complex.[14] However, the currently acceptable classification of CRS (table 1) is based on the agreement in the Consensus Conference of the Acute Dialysis Quality Initiative in 2008, depending upon the "prim moven" of the disease process.[1,7,15]
In summary, CRS is classified into five types (CRS, RCS, and secondary CRS). In type 1 (acute) and type 2 (chronic) CRS, acute or chronic HF can cause AKI, CKD, or failure. AKI causes acute HF in type 3 (acute RCS), whereas CKD causes chronic HF in type 4 acute RCS (cRCS). In type 5 CRS (secondary CRS [CRCS]), systemic illnesses, such as amyloidosis, DM, and hypertension, cause simultaneous acute or chronic kidney disease (CKD) and HF. We propose a revision of this classification and nomenclature based on the organ that is affected first, leading to damage to another organ (i.e., CRS, RCS, and cardio-reno-cardiac syndrome).
Understanding the predisposing or triggering events of kidney-heart interaction syndrome and its natural history is essential for studying the epidemiology and prognosis of CRS. Determining heart–kidney interaction epidemiology by CRS subtype is essential for understanding the illness burden for each subtype, identifying information gaps, and designing future studies.
The 2007 report from the Acute Decompensated Heart Failure National Registry (ADHERE) database analyzed data from 118,465 patients admitted with ADHF. The report found that 9% of patients had normal renal function upon admission. Additionally, 27.4% had mild renal dysfunction (defined as a glomerular filtration rate [GFR] of 60 - 89 mL/min/1.73 m2), 43.5% had moderate renal dysfunction (GFR of 30 to 59 mL/min/1.73 m2), 13.1% had severe renal dysfunction (GFR of 15 to 29 mL/min/1.73 m2), and 7% had a GFR less than 15 mL/min/1.73 m2 or were on chronic dialysis.[16,17] Additional extensive datasets have shown that the frequency of cardiac or renal dysfunction is positively correlated with the occurrence of the other, which implies a considerable challenge in assessing the exact epidemiology of each type alone owing to the interactions between the types of these syndromes.
In ACRS, most of the literature has focused on the examination of AKI caused by deterioration of cardiac function. The vast majority of investigations are conducted using retrospective, secondary, or post hoc analyses of databases, [1,2,18–21] or clinical trials focusing on pharmacological treatment.[22,23] The term worsening of renal function (WRF) is used to refer to rapid or gradual alterations in renal function in individuals with ADHF. The estimated incidence of WRF ranges from 19% to 45%. The wide variety of results may be attributed to differences in the definitions of WRF, the duration of observation, and the population being studied. Most studies have shown that the occurrence of WRF/AKI in ADHF/ACS occurs soon after the patient is admitted to the hospital. ADHF and acute coronary syndrome (ACS) have shown a correlation between the occurrence of WRF/AKI and increased mortality rates, both in the short and long term, as well as extended hospital stays. Additionally, there is an association with higher readmission rates, faster CKD progression, and increased healthcare expenses.[24] Furthermore, it seems that there is a direct correlation between AKI severity and the likelihood of mortality.[25]
Two trials have shown that the risk of an adverse outcome is present regardless of whether WRF or AKI is temporary or chronic.[25] Additionally, even minor acute fluctuations in SCr levels (0.3 mg/dL) may alter the likelihood of mortality.[22] Venous congestion might be a significant hemodynamic component that contributes to WRF in patients with ADHF.[26] Among ADHF patients managed in the intensive care unit, the occurrence of WRF was linked to higher central venous pressure (CVP) both at admission and after intensive medical treatment.
CKD and cCRS frequently co-occur to the extent that it is often impossible to determine which disease occurs first in clinical scenarios. There has been no differentiation between aCRS and cRCS in extensive database studies. Nonetheless, evidence of CKD is present in 45–63.6% of patients with CHF.[16,27] In general, researchers observed a "dose-response" or graded correlation between kidney function decline and unfavorable clinical outcomes. In long-standing congenital heart disease, the kidneys adopt changes in perfusion and stimulation of neurohormonal pathways under specific conditions, delaying the occurrence of cCRS. Over 50% of the 1102 adult congenital heart disease patients in the study had developed kidney dysfunction, and 9% had an estimated (eGFR) of less than 60 mL/min/1.73 m2.[27,28] The mortality rate of the latter cohort increased by a factor of three. Patients with uncomplicated anatomical cardiac defects and congenital heart disease exhibit renal dysfunction at some stages. Additionally, the fact that patients may progress from aCRS to cCRS at different times complicates the task of delineating the epidemiology of aRCS.
aRCS is challenging to define epidemiologically for the following reasons: (A) substantial heterogeneity in precipitating conditions; (B) divergent definitions of acute heart and kidney dysfunction; (C) inconsistent baseline risk for acute cardiac dysfunction development (i.e., increased susceptibility in individuals with subclinical CVD); and (D) omission of acute heart dysfunction in numerous clinical reports of AKI. As a result, acute cardiac dysfunction leading to AKI is primarily characterized by disease- and context-dependent incidence assessments and clinical outcomes. A report deliberated on whether epidemiological studies should employ the RIFLE/AKIN criteria to define AKI.[29]
An instance of aRCS may manifest as arrhythmia, ACS, or AHF following acute glomerulonephritis, acute cortical necrosis, or other AKI-induced illnesses. Elevated electrolyte levels, humoral mediators, toxemia, and sodium and fluid retention are potential contributors to acute cardiac dysfunction. Cardiovascular surgery-associated AKI (CSA-AKI), in which AKI participates in the development of fluid excess and latent heart dysfunction, is an additional example. We acknowledge the possibility that CSA-AKI may constitute aCRS. The distinction between these two manifestations could potentially be significant, given that they might differ substantially in terms of epidemiology, outcomes, risk factors, and the therapeutic interventions required. The reported incidence of CSA-AKI ranges from 0.3% to 29.7%.[30–32] Due to the various definitions in use, this wide variation in incidence exists.[33] Nevertheless, comprehending aRCS epidemiology is difficult because its associated risk factors do not account for the fact that the precipitating event for CSA-AKI may be predominantly HF or AKI-related.
Several observational trials have assessed the CV event rates and prognoses in specific CKD populations for CRCS.[34–38] In comparison to age and gender-matched non-CKD individuals, cardiac-specific mortality rates in CKD patients are 10 to 20 times higher, and cardiac disease is prevalent among this CKD population.[34,39,40]
Multiple observational studies have identified a correlation between the degree of renal dysfunction and increased cardiac event risk, as well as a gradual increase in HF and CVD prevalence.[38,40–43] Similar patterns of mortality from all causes and cardiac-specific causes mirrored this dose-response relationship. [34,37,38,41,44,45] Consequently, CKD probably increases the incidence and progression of CVD.[36,42,43]
Owing to the multitude of potential acute and chronic generalized diseases contributing to CRCS, data on CRCS epidemiology are limited. As a result, the incidence estimates, risk identification, and outcomes associated with CRCS are primarily disease- or context-dependent and may vary over time. Several chronic systemic diseases (e.g., DM, HTN, and amyloidosis) may potentially meet the criteria for CRCS. However, these clinical features may also meet other CRS subtype criteria at specific points in the natural disease course. The pathophysiological mechanisms underlying secondary heart-kidney interactions remain poorly understood.
Sepsis is a prototypical condition that can result in RCS. Mortality estimates ranged from 20% to 60%. Sepsis is prevalent, and its frequency is rising in RCS.[46,47] AKI in sepsis is related to increased associated illness and death,[46–48] affecting 11–64% of septic patients.[49–52] In sepsis, cardiac dysfunction is prevalent.[53–55] Approximately 30–80% of patients with sepsis have raised cardiac-specific troponins,[55–58] which frequently correlates with impaired left ventricular function.[55,57,58] AKI and myocardial injury or dysfunction are incredibly prevalent in severe sepsis or septic shock; however, there is a shortage of epidemiological and integrative studies that have investigated their incidence, risk identification, pathophysiology, and associated outcomes. We intended to make this section slightly longer because we will not give too much detailed epidemiological information while discussing each CRS type separately.
Anemia and Malnutrition
Anemia, cachexia, and dietary deficits often lead to an increase in tumor necrosis factor-alpha (TNF-α) and other pro-inflammatory cytokine levels, which are commonly associated with chronic RF and HF. Ultimately, this leads to further harm and scarring in these and the other body organs.[2] Anemia in CRS and RCS is a pathological triad in which failed kidney and heart function may cause anemia.[59] Anemia may aggravate HF and renal dysfunction, leading to a vicious loop that impacts morbidity and mortality.[60]
The prevalence of anemia ranges from 14% to 70% and increases with chronic HF severity, CKD stage, and age. Treating anemia improves cardiac and renal function and reduces hospitalizations for HF.[61] Most patients with advanced CKD and chronic HF have chronic disease-related anemia, and in chronic HF patients, anemia is common and linked to higher death rates.[62] The optimized HF registry links anemia to a 30% increase in all-cause mortality and morbidity.[63] The Anemia in Chronic Heart Failure: Outcomes and Resource Utilization (ANCHOR study) assessed the influence of CRS and anemia on mortality. A study found that high hemoglobin levels (>17 g/dL) or low hemoglobin levels (<13 g/dL) independently increased the risk of death and hospitalization in CRS patients with impaired or intact systolic function.[60] In anemic chronic HF patients, reduced oxygen delivery to tissues leads to hemodynamic and non-hemodynamic responses, contributing to higher mortality. Anemia responses, such as increased left ventricle workload, RAAS and SNS activation, sodium and water retention, decline of the GFR, and renal blood flow (BF), lead to HF deterioration and adverse outcomes.[64] Anemia is an independent predictor of death in chronic CRS patients, with a frequency ranging from 5% to 55%.[64,65] Around 25% of patients in the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) registry were moderately to severely anemic. In comparison, 51.2% of patients had mild anemia (Hb<12.1 g/dl), and 25% were moderately or severely anemic were studied, and revealed that severe anemia patients had worsened outcomes.[63]
Multiple contributory factors have been recognized for the development of anemia in CRS and RCS patients. Advanced age, low BMI, diabetes, lower LVEF, omission of RAAS inhibitors, and use of intravenous (IV) loop diuretics independently correlated with anemia severity. [60] Anemia in patients with HF can be caused by folate and vitamin B12 deficiencies, iron deficiency, blood loss from aspirin and anticoagulants, increased plasma volume and hemodilution, inflammation, renal insufficiency, poor nutrition, and intestinal malabsorption due to edema.
Furthermore, CKD anemia has several causes, including insufficient EPO synthesis, restricted iron availability, elevated hepcidin levels, decreased EPO receptors, and use of Angiotensin receptor blockers (ARBs) and Angiotensin-converting enzyme inhibitors (ACEi). [64,66] Although chronic inflammation leads to elevated EPO levels in HF patients, the erythropoietic EPO action is ineffective on the bone marrow.[66,67] Additionally, prolonged inflammation increases hepcidin production, limiting iron absorption and bioavailability for hemoglobin formation.[68]
Anemia plays a multifaceted role in the pathophysiology of CRSs. A heart under stress or a kidney injured previously may experience ischemia insults from the Hb reduced oxygen carried ability in anemia, which may lead to progressive cell death in the kidneys and heart.[63,69] Because red blood cells are rich in antioxidants, anemia may lead to an increase in oxidative stress.[70] Anemia may result in peripheral vasodilation, ischemia, RAA, Inactivation, and the release of ADH, causing vasoconstriction, water, salt retention, and persistent renal venous congestion. Persistent chronic venous congestion eventually leads to nephron damage and interstitial fibrosis. In addition to LV enlargement, ischemia and necrosis lead to myocardial cell death in persistent severe anemia.[71,72]
While erythropoiesis-stimulating agents (ESA) can improve anemia in chronic HF patients and improve outcomes and quality of life, normalizing Hb levels may not have the same positive effects. Surprisingly, trials aiming for Hb levels over 13 g/dL were linked to a greater incidence of adverse events on CKD progression and CV death.[73,74] More than 4000 patients participated in a trial to minimize CV events with darbepoetin alfa treatment.[75] Darbepoetin alfa did not lower the mortality risk, CV events, or renal events in patients with DM, CKD, or mild anemia who did not receive dialysis and achieved the targeted Hb levels (approximately 13 g/dl). Individuals who received darbepoetin alfa had a higher risk of fatal or nonfatal stroke. A study named The reduction of events by darbepoetin alfa in heart failure (RED-HF) included 2278 patients with mild to severe anemia (Hb between 9-12g/dl) and systolic HF.[76] According to the study's findings, Darbepoetin alpha did not enhance clinical outcomes in patients with mild-to-moderate anemia and systolic HF. Hence, they concluded that darbepoetin alpha use is advisable in these patients. Further studies are required to confirm this hypothesis.
Anemia is challenging to treat, particularly in individuals with chronic kidney disease (CKD) and congestive heart failure (CRS. Targets based on CKD Hb level recommendations (10–12 g/dl) or higher (12–13 g/dl) but less than 13 g/dl (because studies with Hb levels of 13 g/dl or above were linked with unfavorable results) are still unclear. However, this remains an unresolved issue, and further studies are required. Currently, there are no evidence-based suggestions for the treatment of anemia in patients with chronic HF and CKD. It is necessary to manage anemia, renal insufficiency, and heart failure simultaneously to treat these patients. KDIGO's international conference found that erythropoiesis-stimulating agents (ESAs) could neither prevent nor cure anemia in HF or CKD patients.[77,78] In contrast, in many trials, IV iron therapy for chronic HF patients with iron deficiency, including anemia, eGFR, increased functional capacity, and symptoms.[79] IV iron and ESAs are CKD patients' primary treatment for anemia.[80] ESAs are not advised for patients with anemia because of the unfavorable results of anemia overcorrection, leaving IV iron as the primary treatment. IV iron treatment improves iron parameters, NYHA functional status, and life quality in HF anemic or non-anemic patients or CKD.[79,81,82] ESA treatment may reduce LV thickness and mass and improve renal parameters. [83] Darbepoetin alfa treatment for anemia reported outcomes in mild or severe anemia and systolic HF and may even increase thromboembolic rates. [83] The American College of Cardiology Foundation, Heart Failure Society of America, and European Society of Cardiology advise against using ESAs for anemia management in HF patients.[84] ESA trials in anemic and CKD patients show a greater risk of CV events with higher Hb values.[85,86]. ESA medication is administered to a limited percentage of cRAS patients, following KDIGO guidelines for treating anemia in CKD patients.[87] Furthermore, IV iron administration in HF and CKD is beneficial for patients with HF and anemia; however, overcorrection should be avoided.
Hypoxia-inducible factor prolyl hydroxylase inhibitors (HF-PHIs) (vadadustat, daprodustat, and desidustat) are a new family of medicines used for anemia therapy in patients with CKD and cRAS. These inhibitors increase physiological EPO synthesis by blocking prolyl hydroxylase enzymes, which degrade hypoxia-inducible factors (HIF) and trigger EPO expression in hepatic cells and kidneys. HIFs affect EPO and initiate a coordinated response that increases iron absorption and decreases hepcidin levels, resulting in improved iron mobilization and utilization. Clinical experiments using HIF-PHIs revealed reduced ferritin and hepcidin levels, increased erythropoiesis, and raised overall iron binding capacity.[88] Recent studies on oral HIF-PHIs have shown results in maintaining or improving anemia in CKD patients.[89] HIFs may have adverse impacts on many organs, cellular functioning, angiogenesis, tumor development, and glucose metabolism.[89] HIFs' long-term use of HIFs requires further research.
Obesity
According to research, overweight and obesity-induced glomerulopathy is a disorder where obese persons without DM experience excessive filtration in the kidneys, which eventually leads to CKD and CRS, particularly in cCRS and cRCS.[90] In the absence of severe DM, obesity has been associated with a roughly seven-fold increase in the chance of developing aCRS, especially in individuals with associated clinical conditions.[90,91] Additionally, it has been recognized that adipocytes release IL-6 and TNF-α, which have been linked to fibrosis-induced cardiac and renal disorders.[92]
Hypertension
Elevated blood pressure is known to cause direct damage to the kidneys and heart as well as an increase in the activation of the sympathetic neurohumoral system, which increases heart and kidney stress and ischemia. Furthermore, there is a correlation between this and the rise in the occurrence of renal failure, especially in those with decompensated congestive HF.[6]
Diabetes Mellitus
The recently established cardiometabolic renal syndrome describes the systemic interrelationship of type 2 DM, CVD, and CKD according to expanding data.[93] Approximately 500 million individuals worldwide have DM with the majority having type 2 DM.[94] Approximately 64 million people worldwide have HF [95], and approximately 700 million have CKD [96], which is the biggest pandemic of the 21st century. Patients diagnosed with HF face a heightened prevalence of Type 2 DM (20%) compared to those without HF (4–6%),[97] as well as a corresponding increase in the risk of CVD (two-to-four-fold).[98] Recent studies have shown a CKD prevalence of 40% in type 2 DM[99] and 50% in HF.[100] Conversely, CKD patients had a higher CVD diagnosis rate than the overall population.[101]
Diabetes is well recognized as a contributing factor to glomerular damage and dysfunction, leading to the ultimate loss of functional filtration units and the onset of CKD. Furthermore, both DM and hypertension can lead to damage in the mesangial, podocyte, and endothelial cells, which may cause excessive release of albumin into Bowman's space. As a consequence, the proximal tubular cells experience an increase in the amount of effort they need to reabsorb substances.[6,92] Studies have shown that this process causes programmed cell death in renal tubular cells, an increase in the rate of nephron loss, and the progression of kidney disease. Albuminuria and extensive proteinuria are often observed in cases of severe renal damage in different contexts. The risk factors are summarized in Figure 2.
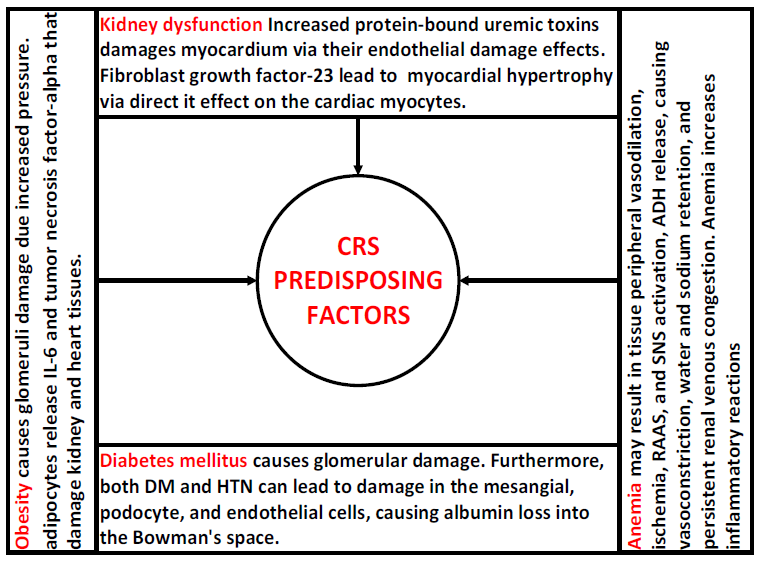
Figure 2. Risk factors for cardiorenal syndrome Types.
Abbreviations: Cardiorenal syndrome (CRS), diabetes mellitus (DM), hypertension (HTN), interleukin-6 (IL-6), renin-angiotensin-aldosterone system (RAAS), and sympathetic nervous system (SNS).
Various mechanisms have been proposed for CRS and RCS.[11,14,102] Generally, the mechanisms responsible for RCS and RCS are summarized as follows: A) increased intra-abdominal and central venous pressure, B) SNS activation, C)hormone-mediated, D) anemia, E) oxidative stress, F) infection and inflammation, and G) decreased cardiac stroke volume. Nonetheless, other mechanisms might exist; for example, in aRCS, the direct and indirect effects of AKI on the heart, and in aCRS, hemodynamic and hemodynamic mechanisms. The generalized mechanisms proposed for CRS, RECS, and CRCS pathogenesis are shown in Figure 3.
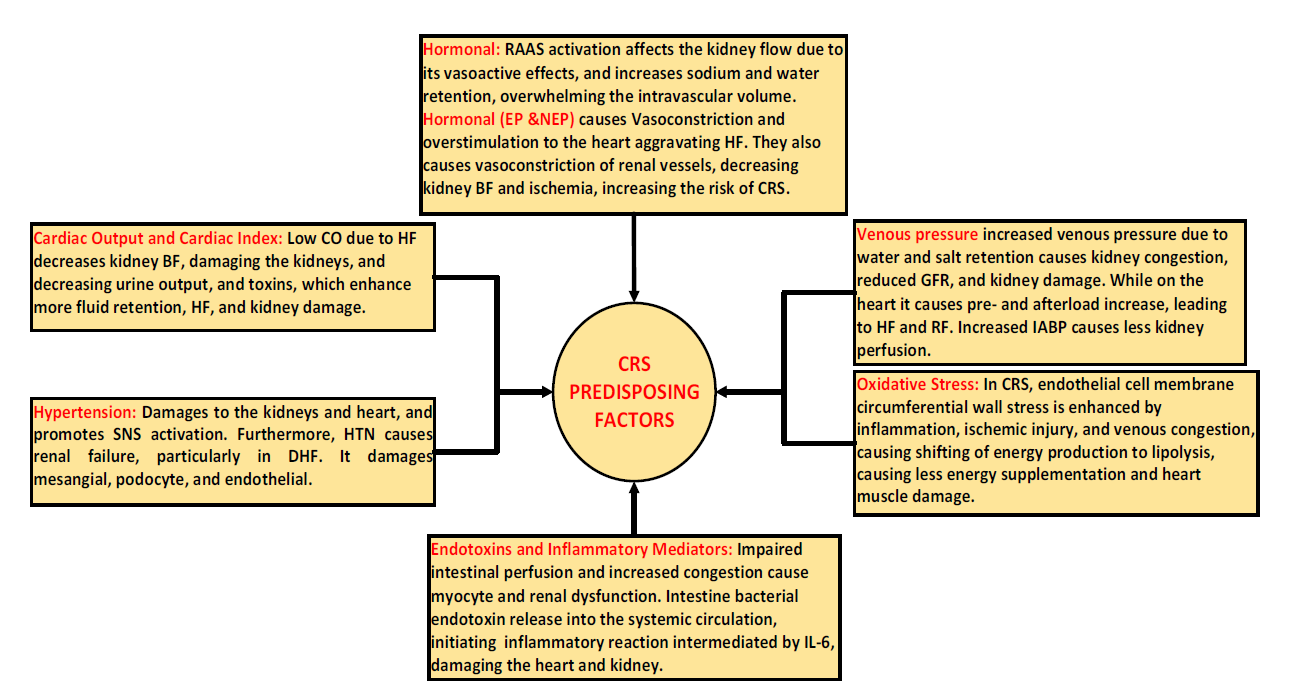
Figure 3. Proposed pathophysiology mechanism of cardiorenal syndrome
Abbreviations: Cardiorenal syndrome (CRS), blood flow (BF), hypertension (HTN), interleukin-6 (IL-6), renin-angiotensin-aldosterone system (RAAS), decompensated heart failure (DHF), heart failure (HF), renal failure (RF), and glomerular filtration rate (GFR).
It was reported that AKI is not consistently associated with acute HF because of the compensatory physiological mechanisms that preserve kidney blood flow,[11,14,102] except if the cardiac output (CO) is reduced significantly.[103] This concept was supported by a post hoc trial analysis that concluded no association between kidney blood flow and cardiac index in congestive HF patients.[104]
Hypervolemia due to sodium and fluid retention is a characteristic feature of acute HF. CO is usually mildly diminished in acute HF cases, and systemic perfusion is usually sufficient to preserve organ perfusion and function. In the cardiogenic shock state, patients may be in normo-, hypo-, or hypervolemic status.[9] These two conditions cause renal injury via distinct mechanisms and have different therapeutic implications. As discussed later, reduced renal perfusion due to renal venous congestion is now believed to be the major hemodynamic mechanism of renal injury in acute HF (aCRS). However, in cardiogenic shock, renal perfusion is reduced due to a critical decline in cardiac pump function.
Cardiac Dysfunction Role
Primary cardiac dysfunction is the principal cause of both aCRS and cCRS. It is difficult to identify the risk factors for AKI or CKD in patients with primary HF because of the common nature of comorbidities and CRS. Certain shared risk factors between the heart and kidneys, such as atherosclerotic risk factors for coronary artery disease, worsen renal function. The primary CRS risk factors are CKD, hypertension (HTN), HF, coronary heart disease, ischemic cardiomyopathy, pulmonary edema, old age, AKI, DM, and pulmonary HTN. In HF, intravascular pressure, CO, and cardiac stroke volume increase SNS stimulation, brain natriuretic peptide (BNP), N-terminal pro-brain natriuretic peptide (pro-BNP) formation, and RAAS activation. These include inflammatory processes, oxidative stress, and other damage to the heart and kidneys.
Baseline SCr independently predicted worsened renal function (WRF) in 299 decompensated systolic HF patients.[105] A retrospective analysis of hospitalized HF patients found that SCr at admission >1.5 mg/dL and a history of chronic HF predicted WRE.[106] The two studies also showed that baseline renal function and HF history strongly predicted WRF. WRF also correlates with CKD risk factors such as DM and HTN.[107] A post hoc analysis of the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE) trial found that prior history of DM and HTN increased SCr by > 0.3 mg/dL, increasing the CRS risk.[106] Old patients and those who have atherosclerosis tend to develop CRS more than others.[2]
Albuminuria with AKI predicts the future HF risk in the general population.[108] Obesity and cachexia may have caused the CRS in this group. Besides hyperfiltration, adipocyte-derived cytokines might damage the kidneys.[109] previous HF and cachexia may cause renal damage and inflammation.[110] Given the substantial relationship between weight and WRF with CKD and heart disease progression, epidemiologic studies have not shown a link with CRS.[111–113]
Finally, CRS patients experience worsening treatment-related kidney dysfunction. Renin-angiotensin-aldosterone system inhibitors (RAASi) and diuretics are essential for treating most heart and renal diseases. The Survival and Ventricular Enlargement trial included 2231 left ventricular dysfunction and acute myocardial infarction. SCr >2.5 mg/dL subjects were eliminated from the Survival and Ventricular Enlargement Trial, which randomly allocated the participants to captopril or placebo between 3 and 16 days (average, 11 days) following acute myocardial infarction. WRF was not linked significantly with captopril use (5.7% versus 6.4%, P = 0.38).[22] A nested case-control study of 382 HF hospitalized patients found no association between RAASi medication usage and WRF.[106] Interestingly, this trial's WRF patients were on larger diuretic dosages.[105,114] In contrast, RAASi may improve chronic HF outcomes, although the SCr level increases at the initial doses, which should not prevent RAASi consumption. ESCAPE trial analysis found no influence on renal outcomes with a loop or total dosage diuretics, although WRF was higher with thiazide diuretics when eGFR was <60 mL/min (P = 0.04).[23] Thiazide is used when loop diuretics fail; hence, it may be related to heart or kidney dysfunction severity. In a post hoc analysis of randomized, open-label research evaluating the effectiveness of continuous with intermittent furosemide in ADHF inpatients, high-dose diuretics (>125 mg/d) were linked with increased in-hospital WRF (P = 0.001).[115] These results reveal a link between increasing diuretic dosage and CRS, but causality is unclear since many processes that cause diuretic resistance in advanced HF might also indicate cardiac severity.[116] Hence, further research is required on this topic.
Cardiac Output and Cardiac Index Role
Initially, it was believed that the significant decrease in kidney function seen in HF was caused by inadequate kidney blood flow due to diminished blood pumping by the heart. Low perfusion pressure or insufficient blood flow to the kidney triggers renin hormone release by juxtaglomerular cells in the afferent glomeruli arterioles. This release is prompted by low flow in the ascending limb of the loop of Henle and pressure-sensing baroreceptors. Increased renin and reduced blood flow to the ascending loop of Henle and afferent arteriole vasoconstriction lead to sodium retention, vascular congestion, and renal dysfunction progression.
Research on animals showed that rats with left ventricular dysfunction due to healed infarcted myocardial reduced their ability to respond to a sudden increase in sodium levels and fluid volume. This study provided a model for understanding circulatory impairment.[117] Theoretically, inotropes may enhance contractility, heart rate, and cardiac index (CI), temporarily enhancing urine production and heart pumping function. Nevertheless, observations indicate that this idea is very restricted, and treating patients with CRS based purely on the low-flow hypothesis does not result in improved results. These findings are supported by the results of the evaluation of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE). This trial compared the use of a pulmonary artery catheter for hemodynamically guided therapy of ADHF with standard clinical care.[118] A study examining 433 people who were hospitalized with ADHF concluded that there was no association between initial kidney function and CI. In addition, enhancing CI did not lead to improved renal function, mortality reduction, or a decrease in the readmission rate. A significant limitation of the ESCAPE study was that patients were omitted if they were in cardiogenic shock or if the investigators believed intrusive hemodynamic monitoring was necessary based on clinical judgment.[118]
In contrast to these results, more recent research conducted on individuals with acute cardiogenic shock did discover a correlation between lower CI and AKI.[119] The findings indicate that in patients experiencing a sudden and significant decrease in CO or a severely reduced CO, a condition of low forward flow pathophysiology leads to CRS. Although this theory suggests that there is a low forward flow, the use of inotropic medications to treat HF and AKI in different patient groups did not impact clinical outcomes. This finding supports the idea that the etiology and management of CRS are more complex than previously believed.[120] Therefore, in an emergency, the impact of CI may vary and be a factor in the most extreme cases of ADHF. However, it is unlikely to function substantially in most patients.
Although these studies have focused on ADHF, there is scarce evidence regarding chronic HF. A single study examined right heart catheterization patients but did not differentiate between the acuity or stability of their HF condition. This study revealed a correlation between renal function and CI. However, it was not the only contributing factor to the hemodynamic process.[121] In addition, the central venous pressure is another significant hemodynamic component that affects renal performance. The significance of these factors as initiators of syndromes is not well understood; hence, new projects are required.
Increased Central Venous Congestion Role
Considering the existing clinical data, attention has recently been directed towards renal venous congestion. As per Poiseuille's law, blood flow through the kidneys is contingent upon the pressure gradient, with high arterial and low venous pressure.[102] In increased central venous pressure in stable HF, elevated renal venous pressure diminishes renal perfusion pressure, thereby impacting renal perfusion and GFR. This is now acknowledged as a significant hemodynamic mechanism of aCRS development.[9] Interestingly, it was reported that CVP of more than 6 mmHg was linked with WRF and an increase in the death rate in CVD patients.[8], and WRF are more often associated with increased intrabdominal pressure.[122] Moreover, Regarding CHF, a negative correlation exists between GFR and increased CVP.[92,123] An increase in CVP results in a concomitant increase in the renal venous pressure and renal interstitial hydrostatic pressure. Hence, renal dysfunction may ensue when the interstitial hydrostatic pressure exceeds the tubular hydrostatic pressure, leading to tubule collapse and a significant reduction in the net ultrafiltration pressure.[92,124]. Furthermore, renal congestion might indirectly affect renal function. This may lead to congestion and swelling in the renal interstitium, which can subsequently increase the pressure inside the renal tubules, resulting in a decrease in the pressure gradient across the glomerulus,[125] impairing kidney function. Additional significant indications of systemic congestion include intestinal and splanchnic congestion, which causes intestinal edema and, less often, ascites. These aggravate the already elevated intra-abdominal pressure, which exerts pressure on the kidney vascular system, impairing blood flow to the kidneys.[126] Hence, paracentesis and systemic decongestion may, expectedly, relieve increased intra-abdominal pressure and improve blood flow to the kidney. Further research is required to confirm this hypothesis.
Alternatively, hypovolemia-induced AKI is the primary differential diagnosis that must be considered in suspected aCRS cases. Patients with stable HF often have a slight excess fluid volume in their bodies (mild hypervolemia). However, they may be fluid-depleted (hypovolemia) due to excessive diuretic use, severe diarrhea, or other factors. Despite the contrasting fluid statuses of patients with aCRS and hypovolemic AKI, distinguishing between them might be challenging. Urine electrolyte levels indicated pre-renal acute kidney damage in both situations. Recent fluid losses or excessive use of diuretics might assist in identifying hypovolemia.
Moreover, body weight change, if accessible, may be crucial for accurately determining the correct diagnosis. Misdiagnosis of aCRS as hypovolemia-induced AKI may have disastrous consequences. Incorrect attribution of AKI to volume depletion may exacerbate both cardiac and renal function if fluid is administered, further sustaining the vicious cycle. If renal function does not improve, additional fluid may be administered. Hence, awareness of these issues among health professionals is essential to improving outcomes.
Right Ventricle Dilatation and Dysfunction
Left ventricular (LV) filling is impaired, and forward output results due to right ventricle (RV) dilation, which operates through a ventricular interdependent effect (referred to as the reverse Bernheim phenomenon or reverse Bernheim syndrome).[127] Elevated pressure within a distended RV raises LV extramural pressure, decreasing LV transmural pressure about intracavitary LV pressure. This results in the induction of leftward interventricular septal bowing, which further reduces LV preload and distensibility, CO, and forward flow.[128,129] Experimental observations indicate that while an intact pericardium contributes to ventricular interaction, it is not essential.[130] Additionally, when the RV pressure rises, it causes a higher venous circulation pressure and a decrease in CO and BF, negatively affecting heart and kidney function. Therefore, decreased RV filling pressure may increase GFR when treating HF. Reducing renal venous pressure also mitigates the interdependent impairment of left ventricular filling.[131]
Renal Dysfunction Role
Protein-bound uremic toxins (PBUT) are gaining interest because they are associated with CVD [132]. Indoxyl sulphate (IS) and p-crestyl sulphate (PCS) are the two most thoroughly investigated PBUTs involved in the development and advancement of CRS. Both substances were eliminated by tubular secretion in healthy kidneys. Experimental investigations have shown that IS and PCS have a harmful impact by altering oxidative stress, impairing endothelial function, and promoting atherosclerosis. Both substances have been linked to kidney damage, reduced growth of endothelial cells, and hindered healing of wounds, indicating their involvement in tissue damage and possibly in the advancement of CKD.[133,134] An investigation using a nephrectomized animal model revealed PCS's impacts on cardiac cells, which included heightened apoptosis, amplified perivascular and interstitial fibrosis, and a decline in left ventricular diastolic performance. Oxidative stress was identified as a factor in the cardiac muscle alterations generated by PCS.[135] In addition, IS also increases oxidative stress in the kidneys and heart, resulting in cardiorenal fibrosis.[136,137] An analysis of 139 individuals with CKD revealed that indoxyl sulfate was a robust indicator of overall mortality and CV mortality, even after accounting for other factors that may influence the results.[138] While there is evidence indicating a detrimental impact of PBUT on vascular cells, the heart, and the kidney. Hence, further studies are required to gain a more comprehensive understanding of the function of PBUTs in CRS and RCS pathophysiology.
Fibroblast growth factor-23 (FGF23), a hormone produced in the bone, regulates the metabolism of vitamin D and phosphate in the kidneys. It is a reliable indicator of adverse CV outcomes in patients with CKD and ESRD. An increased level of FGF23 has been linked to left ventricular hypertrophy (LVH) and higher death rates in individuals with advanced CKD.[139] There is an ongoing debate regarding whether FGF23 causes myocardial hypertrophy by directly affecting cardiac myocytes. This is because alpha-klotho receptors, responsible for mediating FGF23's actions, are absent in the bones, heart, or other organs. FGF23, a hormone derived from bone, boosts phosphate excretion and reduces 1,25(OH)2 vitamin D production in the kidney by interacting with fibroblast growth factor receptors (FGFRs) activated by the transmembrane protein Klotho.[140] Meta-analyses of conventional epidemiological studies have uncovered a link between elevated levels of FGF-23 and a heightened risk for atherosclerotic cardiovascular diseases, including heart failure and stroke.[141] Additional evidence indicates that FGF23 may directly decrease the ability of the heart muscles to contract and relax, promote the growth of heart muscle cells, and raise the likelihood of abnormal heart rhythms via changing the intra- and extracellular calcium movement.[142] In the kidneys, FGF23 reduces phosphate reabsorption by decreasing the expression of sodium-phosphate cotransporters inhibitors in proximal tubules.[143] In summary, alpha-Klotho has the potential to serve not only as a predictive or prognostic biomarker for acute or progressive kidney disease but also as a therapeutic agent to mitigate kidney damage, promote kidney recovery, and reduce the risk of acute or CKD. [140]
Sympathetic Nervous System Activation
Effects of Sympathetic Nervous System Activation on The Heart in Cardiorenal Syndrome
Acute HF may decrease CO via different causes, such as atrial fibrillation, other arrhythmias, post-myocardial infarction, or any other cardiac or non-cardiac diseases. However, decreased CO has no significant role in the AKI pathogenesis in acute HF.[9]
In HF, SNS becomes more active and extensive if CO progressively decreases. Its activation causes afferent arterioles vasoconstriction and efferent vasodilation, reducing renal perfusion and increasing tubular sodium and water reabsorption.[14] SNS activation occurs in response to receptor stimulation in different parts of the CVS. As these receptors are stimulated, different reflexes are produced. Borovac et al. well illustrate the pathophysiological implications of SNS activation in HF.[144]
Under physiological circumstances, complex autonomic CV reflex integration regulates the degree of SNS activation and sympathetic outflow to the peripheral circulation and heart. The reflexes consist of the following: cardiac chemo, pulmonary stretch, peripheral and central chemoreceptor reflexes, cardio-cardiac reflexes, cardiopulmonary mechanosensitive reflexes, arterial baroreflexes, and afferent projected reflexes from skeletal muscles.[145] Each reflexes function similarly to regulate and sustain vascular tone, mean arterial blood pressure, ventilation, heart rate, and respiratory drive in reaction to diverse hemodynamic fluctuations due to sympathetic/ parasympathetic autonomic system imbalance.[146]
Impaired baroreceptor and chemoreceptor responses, increased release of catecholamines in the bloodstream and neurons, reduced parasympathetic response, and heightened sympathetic activity towards the heart and kidneys were observed in HF. When these persistent sympathoexcitatory effects on the cardiovascular system occur over an extended period, they start a harmful cycle of HF progression. The damages are linked to the heart muscle cells' apoptosis, unfavorable changes in the structure of the heart and blood vessels, and arrhythmia.[144] In HF, the decreased baroreceptor reflexes lead to excessive SNS activity, elevating renin release from the juxtamedullary cells of the kidneys and augmenting the vasoconstriction effect.[147] Thus, it is essential for physicians to accurately diagnose and characterize the extent of increased SNS activity in patients with HF to prevent renal failure and CRS development. It is important to prioritize using neurohumoral antagonists to treat these conditions effectively, reduce their negative consequences, and enhance the overall results. The supplementary use of sophisticated imaging techniques and innovative biomarkers might assist in the process of making clinical decisions.[144]
The activation of the SNS is a fundamental physiological reaction to stressful situations such as low blood volume, low blood sugar, low oxygen levels, or heart problems.[148] SNS activity can alter and cause a broad range of powerful effects on blood flow, including increasing heart rate (positive chronotropic effect), an increase in cardiac muscle contraction strength (positive inotropic effect), an increase in relaxation of the heart (positive lusitropy), improving the conduction of electrical signals between the atria and ventricles (positive dromotropy), decreasing the capacity of veins to hold blood, and constricting the blood vessels in the periphery and skin, trying to improve cardiac stroke and CO.[149,150] The SNS effect primarily relies on increased norepinephrine release from SNS terminal neurons. In addition to SNS terminal secretion of epinephrine, the adrenal medulla also secretes a significant amount of epinephrine as a part of SNS activation.[144]
Additionally, peripheral vasoconstriction due to SNS stimulation preserves the mean arterial perfusion pressure and accelerates HF progression. Moreover, the withdrawal of normal restraints that influence excitatory inputs increases and stimulates central integratory sites, which have been linked to SNS activation. Dysregulation of the CVS beta-1 adrenergic receptor (ADRB1) signaling, and transduction is a fundamental characteristic of HF progression. In contrast, alpha-1-ADRRs and ADRB2s in the heart may operate compensatively to preserve cardiac inotropy. Polymorphisms of adrenergic receptors might influence the pharmacological responses, susceptibilities, and adaptive mechanisms of SNS.[151] Furthermore, Neuropeptide Y,[152] galanin,[153] endothelin (ET-1,2,3),[154] catestatin[155] plasma levels increase. All these mediators and SNSs shared in CRS pathogenesis are not completely understood, and further research is required.
In addition to the effect of SNS activation on the failed heart, it also negatively affects the kidneys. SNS innervation is vital for regulating kidney function. SNS activation causes vasoconstriction of the renal artery and reduces blood flow in the kidney. In addition, SNS induced juxtaglomerular cell renin release and RAAS activation. This boosts blood pressure and salt absorption. Chronic sympathetic overactivity can lead to hypertension (HTN). Drug-resistant HTN develops because of inappropriate SNS activation. Renal denervation (RDN) is used to treat drug resistant HTN via neuromodulation. However, the benefit of RDN was supported by small randomized studies that reported reductions in office and ambulatory blood pressure readings.[156] Nevertheless, the extent of the blood pressure decrease after RDN varies among these studies.[157] Given that RDN is an invasive and costly intervention, there is growing interest in selecting appropriate patients for this procedure. Moreover, some groups of physicians and patients are interested in using RDN to achieve sustained long-term blood pressure control, with the hope that maintaining lower blood pressure levels could be achieved with a reduced dosage of antihypertensive medications.[157]
Effects of Sympathetic Nervous System Activation on The Kidneys in Cardiorenal Syndromes
Furthermore, besides raising blood pressure and activating the RAAS, SNS activation is believed to cause renal damage via additional pathways. Laboratory research found that RDN reduces the severity of renal fibrosis after unilateral ureteral blockage.[158] Administering norepinephrine directly to kidneys that have lost their nerve supply leads to an increase in the production of transforming growth factor-β1 (TGF-β1) and the expression of α-smooth muscle actin (α-SMA) in the interstitial area, leading to excessive accumulation of extracellular collagen matrix. Comparable results were seen in an alternative animal ischemia/reperfusion injury (IRI) model.[158] The tubulointerstitial fibrosis that occurs 4-16 days after the damage caused by IRI is reduced by RDN when performed at the time of injury or within 1day after the injury. Administering calcitonin gene-related peptides produced by afferent nerves or norepinephrine derived from efferent nerves to the kidneys that have been denervated or subjected to ischemia-reperfusion injury (IRI) replicates the effects of innervation.
Unlike HTN and chronic interstitial fibrosis in the kidneys, animal investigations have shown that SNS stimulation protects against AKI. Formoterol, a selective and long-acting agonist of the β2-adrenergic receptor, was shown to be a potent stimulator of mitochondrial biogenesis in the kidney.[159] Formoterol is effective in restoring mitochondrial and kidney function in renal IRI. The results demonstrated that therapy with formoterol successfully recovered renal function, saved renal tubular epithelial damage, and boosted mitochondrial biogenesis.[160] The impact of sympathetic signaling on macrophages in lipopolysaccharide-induced sepsis and the renal IRI model was conducted.[161] Analysis conducted in a laboratory setting showed that ADRB2 stimulation in macrophages led to T-cell immunoglobulin and mucin domain 3 (Tim3), associated with anti-inflammatory changes in their characteristics. β2 stimulation by salbutamol reduces lipopolysaccharide-induced inflammatory responses in vivo. Furthermore, salbutamol administration effectively decreased renal damage induced by IRI. However, these beneficial effects were nullified in mice with deletion of the Adrb2 gene in macrophages. In contrast, salbutamol-treated macrophages were successfully protected from renal IRI. Moreover, single-cell RNA sequencing utilization proved that this safeguarding effect was linked to the buildup of macrophages expressing Tim3 in the kidney tissue.[161,162]
Acute CRS, characterized by the occurrence of AKI accompanied by ADHF, is recognized to be linked with elevated rates of death and hospitalization in HF.[163] In CRS type 2, prolonged heart failure leads to renal damage, resulting in the development of CKD.[164] During a mice study, a condition called non-ischemic hypertrophic congestive HF was induced using transverse aortic constriction (TAC). After 12 weeks, mice showed higher levels of serum creatinine and increased expression of a protein called kidney injury marker 1 (KIM-1). In addition, the kidneys exhibited perivascular and focal tubulointerstitial fibrosis.
SNS activation is believed to play a role in kidney damage in both acute and chronic HF. SNS stimulation appears to have both positive and negative effects on renal fibrosis and AKI development. Nevertheless, little is known about the potential beneficial effects of SN stimulation in CRS and RCS. To elucidate the function of SNS activity in aCRS, an animal study using a combination of TAC and unilateral renal IRI models was performed.[165] The assessment of immediate (within 24 hours) and prolonged (after 2 weeks) stages of kidney damage 8 weeks after thoracic aortic constriction surgery demonstrated that there was no noticeable effect during the immediate renal IRI phase. However, the progression of kidney fibrosis during the prolonged phase was significantly reduced by pre-existing HF. It was reported that conducting RDN two days before IRI induction prevented the reduction of renal fibrosis in TAC animals. Thus, it may be inferred that the safeguarding effect of previous HF on chronic kidney interstitial fibrosis is facilitated by SNS stimulation. While sympathetic activation is thought to play a role in improving kidney recovery after IRI caused by excess pressure on the heart, it is important to mention that a reduction in renal fibrosis associated with previous HF has been reported in mice with renal sympathetic denervation following transverse aortic constriction. These findings imply that factors other than sympathetic stimulation are implicated.[165] This conclusion requires further investigation.
Hormonal Response Role in Cardiorenal Syndromes
The blood pressure detected at the glomerular afferent arterioles and the decreased amount of chloride given to the macula densa also affects renin synthesis.[166] A rise in renin levels results in increased synthesis of angiotensin II (Ang II), which has many detrimental effects on the heart, blood vessels, and kidneys. Ang II induces vasoconstriction in the kidneys' efferent arterioles, leading to a higher proportion of renal plasma filtered through the glomerulus. This leads to an elevation in the oncotic pressure around the tubules and a decrease in the hydrostatic pressure, resulting in intensified sodium reabsorption in the proximal convoluted tubules. Ang II directly stimulates the sodium-bicarbonate co-transporters and apical sodium hydrogen exchangers in the proximal convoluted tubules, increasing solutes and fluid reabsorption.[167] Ang II furthermore stimulates the aldosterone-induced sodium reabsorption in the distal tubules and enhances the expression of endothelin-1 (ET-1) in the kidney.[166,168] ET-1 is a powerful peptide that triggers vasoconstriction, promotes inflammation and fibrosis, and ultimately leads to kidney damage.[169]
Angiotensin II type 1 receptors (AT1) are present in the heart. In animal models, activation of AT1 receptors leads to an increase in the size of cardiac muscle cells (cardiac myocyte hypertrophy) due to the release of transforming growth factor-β1 and ET-1 from the cardiac fibroblast in a paracrine manner.[170] Angiotensin II induces constriction of vascular smooth muscle via the activation of AT1 receptors. In addition, Ang II facilitates oxidative stress by promoting the creation of reactive oxygen species in the heart and kidney tissue, resulting in inflammation and HTN.[171] In individuals with HF, impaired function of the left ventricle leads to activation of the SNS as a mechanism to sustain blood flow. This activation occurs via many mechanisms, including heightened contractility, increased lusitropic (force of contraction), and systemic vasoconstriction.[172] Hence, RAAS activation via Ag II significantly affects water and salt retention and kidney BF, deteriorating renal and heart damage and dysfunction.
Adenosine is secreted in the presence of excess sodium in the distal tubule. It acts on adenosine type 1 receptors located in PCT and afferent arterioles. Adenosine causes constriction of the afferent arterioles, leading to decreased renal blood flow and GFR. In addition, activating adenosine type 2 receptors stimulates the secretion of renin and increases sodium reabsorption by the PCT, decreasing urine production.[173] The effectiveness of adenosine type 1 receptor antagonists in CRS is debatable. The PROstate TEsting for Cancer and Treatment (PROTEC) trial, which studied rolofylline (a selective A1 adenosine receptor antagonist) in patients hospitalized with ADHF, found that the rolofylline group did not achieve the primary outcome of improved dyspnea or secondary outcomes (death, CV events, rehospitalization, or chronic renal impairment). Therefore, more clinical investigations are required to assess the effectiveness of adenosine A1 receptor antagonists in the CRS population.[174]
Antidiuretic hormones (ADH) influence arterial blood pressure and glomerular hemodynamic mechanisms. Patients with ADHF frequently experience increased ADH release. ADH induces water retention through vasopressin V2 receptors in the collecting duct. Studies have shown that increased ADH levels are a factor in CKD progression.[11,175,176] ADH-induced renal hemodynamic effects may be attributable to its influence on the RAAS in the context of the vicious cycling effect. By activating V2 receptors or decreasing sodium concentration at the macula densa, ADH may stimulate renin secretion directly or indirectly.[11,176] There is evidence that patients with LV dysfunction who do not exhibit overt clinical HF have an elevated plasma ADH, which is associated with unfavorable outcomes.[177] Therefore, hormonal and neural responses to changes in AHF and AKI-associated conditions lead to fluid and salt changes that significantly affect intra- and extravascular fluid content, adversely affecting cardiac and kidney tissues.
Serum Chloride Role in Cardiorenal Syndromes
Recent studies indicate sodium chloride might be a cardiovascular and renal health biomarker.[178] Chloride, in conjunction with salt, helps regulate the osmolarity of blood serum, maintain proper fluid levels, balance acidity and alkalinity, and interact with serum bicarbonate. Sodium chloride (NCC) and sodium chloride-potassium co-transporters (NKCC) are kidney-specific transporters that mediate apical NaCl reabsorption in the thick ascending limb and the distal convoluted tubules.[179,180] Decreased chloride levels in the blood stimulate NKCC and NCC in the thick ascending limb of the loop of Henle and distal convoluted tubule, increasing sodium, chloride, and potassium reabsorption.[181] Hypochloremia leads to diuretic resistance, which is an important factor in the progression and management of CRS. Serum chloride level reduction is a crucial process that leads to resistance to diuretics and the activation of neurohormones.[182] Furthermore, there was a strong correlation between low serum chloride levels and inadequate decongestion in AHF. This emphasizes the role of chloride in the kidneys' ability to enhance and regulate salt levels.[183] Chloride inhibits the release of renin, whereas low chloride levels enhance its excretion by activating COX-2 and prostaglandins. This connection is linked to heart failure, resistance to diuretics, and cardiorenal syndrome.[184] Hypochloremia leads to renal vasoconstriction and a reduction in GFR without affecting renal innervation.[185] Hypochloremia in patients with congestive HF is a notable prognostic feature that is associated with increased mortality risk.[186] A study revealed an independent and negative correlation between serum chloride levels and long-term mortality. Curiously, the impact of hyponatremia on prognosis was diminished when chloride levels were within the normal range.[187]
Oxidative Stress Role in Cardiorenal Syndromes
Oxidative stress is defined as an imbalance between antioxidants and oxidants that causes an inordinate accumulation of the former, which then causes cellular damage.[188] Cellular metabolism generates reactive oxygen species (ROS) as byproducts prominently within the mitochondria.[189] Oxidative stress occurs when the generation of ROS exceeds the body's capacity to counteract antioxidative mechanisms. This accumulation of ROS causes endothelial dysfunction, cellular damage, and the advancement of atherosclerosis.
In CRS, endothelial cell membrane circumferential wall stress is enhanced by inflammation, ischemic injury, and venous congestion, which can all induce oxidative stress.[190] Fatty acid (FA) oxidation is the primary cause of heart adenosine triphosphate (ATP) production. However, in the presence of HF, myocytes switch from FA oxidation to glycolysis, resulting in 30–40% reduction in ATP production. In HF, glycolysis compensates for energy deficiency; however, this is insufficient to satisfy energy demands, resulting in a low hypoxemia threshold, cell death, and apoptosis. Furthermore, free fatty acid accumulation in myocytes results from decreased FA oxidation by mitochondria, which results in lipotoxicity.[11,191] A cohort study of patients initially admitted with ADHF but later developed AKI was examined for oxidative stress biomarkers, including endogenous peroxidase, myeloperoxidase, IL-6, nitric oxide, and copper/zinc superoxide dismutase. According to the findings, CRS type 1 Patients exhibited markedly elevated oxidative stress markers.[191]
Furthermore, the activation of SNS and RAAS contributes to the harmful consequences of increased fluid volume and changes in blood flow and significantly enhances the oxidative stress experienced by patients with HF and CKD. Ang II has a harmful impact by stimulating NADPH-oxidase, leading to oxidative damage via ROS production and impairing mitochondrial function.[192] Endothelial cells, cardiac myocytes, and renal tubular cells have shown increased NADPH-oxidase activity.[193]
In addition, certain factors, such as dialysate solution use and uremic toxins, contribute to increased proinflammatory cytokine release and formation, oxidative stress, immune system dysregulation, increased carotid artery intima-media thickness, and hypertrophy of the left ventricle in patients with advanced CKD and end-stage renal disease (ESRD). More than 66% of ESRD patients exhibit increased CV morbidity and death, which conventional cardiac risk factors can explain. Consequently, endothelial dysfunction, hyperhomocysteinemia, and oxidative stress may be contributing factors in the development of these conditions.[11,194]
Endotoxins and Inflammatory Mediators' Role
A state of increased and persistent inflammation characterizes CKD and HF. Inflammation leads to the production of pro-inflammatory biomarkers, which are molecules that promote inflammation. Furthermore, impaired intestinal perfusion and increased congestion result from kidney and cardiac dysfunction, leading to the potential worsening of myocytes and renal dysfunction. This can cause bacterial endotoxin of the intestine to be released into the systemic circulation, triggering circulating immune cells' activation via the secretion of cytokines like interleukin-6 (IL-6), which are comparable in nature to TNF-α.[6,92,195]
These inflammatory biomarkers are essential contributors to the damage and destruction of tissues in both the kidneys and heart, leading to cell death and the formation of fibrous tissue. The inflammatory cascade is initiated and propagated by important triggers such as SNS and RAAS stimulation, venous congestion, ischemia, and oxidative stress. Proteins with inflammatory properties, such as TNF-α and related weak inducers of apoptosis (TWEAK), which are part of the interleukin-1 (IL-1) family and IL-6, have been associated with HF and CKD. TNF-α and IL-6 in the kidneys stimulate the buildup of inflammatory cells in the interstitium by enhancing the production of monocyte chemoattractant proteins. TNF-α induces glomerular injury via mesangial cell death.[196] Biomarkers such as soluble ST2, which belongs to the IL-1 family, may be used to predict the likelihood of death from any cause in HF patients.[197] Analogously, there is a strong correlation between IL-6 levels and the course of illness in CKD. Additionally, IL-6 may be used as a death predictor in CKD patients.[198] Research has shown that individuals with CKD[199] and those undergoing dialysis have elevated levels of these pro-inflammatory markers.[200]
Raised C-reactive protein (CRP) levels play a crucial role in the development of atherosclerosis through several processes. CRP triggers complement system activation and is found in many locations within the first stages of atherosclerotic lesions.[201] CRP dramatically enhances the generation of tissue factors by monocytes, which are a powerful procoagulant. This action is further intensified when inflammatory mediators are present.[202] In research including 4269 persons hospitalized with ADHF, patients with CRP levels in the fourth quartile (≥ 9.6 mg/L) had a significantly greater risk of all-cause death within 120 days after discharge. The association between CRP levels and mortality remained significant even after adjusting for other factors.[203] Elevated CRP values in patients undergoing hemodialysis are indicative of the presence of cardiac hypertrophy, left ventricular dysfunction, and increased risk of death.[204] These inflammatory proteins are passive indicators of disease activity and have an active and intricate function in CRS pathophysiology.[11]
The prevalence of CRS continues to increase worldwide, necessitating an enhanced comprehensive understanding and management of the disease. To reduce the occurrence of CRS and RCS, it is necessary to thoroughly investigate, assess, and treat CRS. To determine this, it is necessary to examine the predisposing factors contributing to CRS's development.[6]
The patient's history and clinical examination assist in distinguishing between acute, chronic, and cardiac or renal decompensation. Helpful historical information includes an acute heart ischemic event that causes severe myocardial dysfunction and renal injury or a recent history of severe vomiting or diarrhea. A history of kidney-damaging drugs and creatinine levels is also valuable.[9]
Clinical examination is usually not helpful in distinguishing between CRS types. Yet, many patients exhibit volume overload symptoms, such as high jugular venous pressure, edema, pleural effusion, ascites, and lung basal crepitation.[9] Hypotension, tiredness, reduced peripheral pulses, and irregular heart rhythms may also indicate decreased CO. Paleness, skin color changes, scratch marks, oliguria, or anuria before cardiac failure may indicate a renal etiology of CRS.
The initial laboratory tests should include a complete blood picture, urea and electrolytes, and urine studies (microscopy, urine sodium, urine protein to creatinine ratio, BNP, troponin, and eGFR). Blood and urine cultures, ANA, serum C3 and C4 levels, anti-double-stranded DNA, and procalcitonin may help patients with CRS type 5. The first assessment should involve an electrocardiogram (ECG) and cardiac monitoring to detect any arrhythmias causing or arising from CRS. Transthoracic echocardiography helps detect pericardial effusion, wall motion abnormalities, and LV ejection fraction (EF). Renal ultrasounds were used to assess kidney size. Smaller kidneys and higher renal echogenicity indicate CKD.[205]
A meta-analysis of 22 studies review concluded that the features most strongly suggested AHF were: In a stable patient, a presence of exertional dyspnea or paroxysmal nocturnal history dyspnea, third heart sound, and chest radiographic evidence of pulmonary venous congestion strongly suggested acute HF.[206] The diagnosis of AHF may be difficult in patients who do not exhibit any of these characteristic clinical signs. An instance of pulmonary vascular remodeling in CHF may render pulmonary edema non-existent despite extremely elevated left-sided pressures.[207] Although pulmonary artery catheterization can guide therapy by revealing elevated cardiac filling pressures, clinical evidence opposes its routine application.[118] Because hemodynamic disturbances observed in acute CRS diminish renal perfusion, urine electrolytes (urea fractional excretion < 35% and sodium fractional excretion < 1%) frequently indicate a prerenal form of AKI. Recent studies have demonstrated that cell-cycle arrest biomarkers, including tissue inhibitors of metalloproteinase 2 and urine insulin-like growth factor-binding protein 7, can identify patients with acute HF at risk of developing acute CRS.[208]
Biological markers provide valuable information about the pathogenic processes involved in CRS, allowing for the early and accurate diagnosis of CRS based on established clinical findings. Research indicates that natriuretic peptides are the most well-recognized biomarkers. These biomarkers serve as the foundation for diagnosis, therapy, and outcomes.[209] Diagnosing renal failure during the early stages is challenging or almost impossible using conventional indicators, such as serum creatinine (Scr). However, there have been ongoing attempts to identify potential markers for the early identification of AKI. Kidney failure patients have a greater incidence of morbidity and death related to CVD, which can be adjusted based on cardiac biomarkers.[210] Natriuretic peptides and several other newly discovered biomarkers serve as cardiac biomarkers and have been proven essential in CRS diagnosis, treatment, and outcome prediction.[211] Furthermore, there is evidence that the CRS-linked biomarkers have a role in the pathophysiologic mechanism of CRS and RCS.[212]
Genes in Cardiorenal Syndromes
A total of 119 CRS genes were obtained from the literature to build a Protein-protein Interaction Network (PPIN). PPIN analysis is a commonly used technique for investigating the contextual function of targeted proteins, predicting new disease genes, functional modules, and illnesses, or identifying novel therapeutic targets. The PPIN-based analysis utilizes both context-specific and generic networks. Further analysis of the modules was done to identify 12 crucial genes inside the network.[209] By constructing a protein-protein interaction network and analyzing it at various levels using the MCODE analytic program, 12 promising genes were identified. Gene ontology enrichment, transcription factor analysis, and pathway enrichment were conducted to understand these genes comprehensively. The discovered genes were determined to be functionally associated with protease binding, modulation of blood vessel diameter, advanced glycation end products-receptor for advanced glycation end products (AGE-RAGE) signaling pathway in diabetics,[213] and the hypoxia-inducible factor (HIF)-1 signaling pathway.[209] Novel potential biomarkers are constantly being discovered rapidly, especially with the newly introduced genomic and proteomic approaches.[214] Therefore, gene identification might help predict CRS outcomes and direct CRS treatment.[110] Hence, it is crucial to identify specific genes within the pathways that are connected to CRS. These findings may provide insights for future exploration of the processes underlying CRS and for formulating possible new therapeutic strategies.
In 2021, Ahmed et al. identified 12 crucial genes in a network using module analysis. These genes can potentially serve as valuable tools for studying pathophysiological processes involved in the early diagnosis of CRS. This research has developed an intricate and powerful mRNA regulatory network associated with CRS, leading to a comprehensive understanding of molecular processes and offering crucial insights for exploring new treatment approaches for CRS. Therefore, experimental validation is necessary to confirm these results. A computational system-based approach offers a methodological framework for identifying potential connections between a single candidate biomarker and the functional interdependence of clinically connected disorders.
Effectively managing the enormous quantity of data generated during clinical investigations involving the heart and kidneys requires a standardized data curation pipeline. Utilizing the Enrichr database for Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, this pipeline will aid scientists in efficiently extracting biomarkers from current literature. Furthermore, it will improve our understanding of genes' processes, functions, and participation. Ahmed et al. reported that a thorough understanding of the molecular characteristics associated with the functional interaction between the renal and CV systems is essential.[209] Studying these advances is necessary to provide new evidence for understanding CRS pathogenesis and prevention.
Ontology is a branch of philosophy concerned with the study of existence, including determining whether objects exist and categorizing different forms of existence. Ontological studies reveal that they possess a high abundance of molecular function protease binding and endo-peptidase inhibitor activities.[209] Hence, these data contribute to expanding our understanding of CRS, enhancing the effectiveness of CRS treatment.
Mining literature-based data has identified significant genes that can serve as CRS biomarkers. However, further investigation of these genes, including gene ontology, pathway enrichment, and complicated protein-protein interaction analysis, is needed.
Renal Tubule Damage Biomarker in Cardiorenal Syndromes
Urine microscopy may help differentiate intrinsic AKI from functional Scr alterations in patients with AHF. In addition, a urine sediment severity score based on renal tubular epithelial cells and granular casts predicted worsening AKI during the hospital stay.[215] Novel urine biomarkers may indicate tubular damage in AKI, and several tests are available for in vitro application.
The 25-kDa protein, neutrophil gelatinase-associated lipocalin (NGAL) in neutrophil granules, released by the renal tubular epithelium, cardiac cells, and other organ locations, has been intensively researched in CRS and RCS and has diagnostic and prognostic relevance in AHF and chronic HF. Renal NGAL is the most highly elevated protein in patients with AKI. Ten trials on almost 2000 CRS patients' meta-analysis found that early raised blood and urine NGAL levels are good predictors of dialysis and mortality.[216] Successive NGAL measurements indicate the predictive significance of AKI in AHF improvement.[217]
Tissue inhibitor metalloproteinase-2 (TIMP-2) and insulin-like growth factor-binding protein 7 (IGFBP7) are biomarkers for tubular damage implicated in G1 cell cycle arrest following early cell damage. Kashani et al. compared TIMP-2 and IGFBP7 with other AKI biomarkers in 728 seriously ill patients without AKI.[218] This research found that urine IGFBP7 and TIMP-2 more efficiently detect AKI than other AKI indicators (P<0.002). TIMP-2 and IGFBP7 have been verified in several AKI situations; however, the relationship between cell cycle block markers is not typical of CRS, and there have been no serial studies on this biomarker combination in AHF. Additional possible tubular damage markers in AKI and their role in CRS or RCS will hopefully be available soon.[15]
BNP and NT-proBNP, urinary biomarkers that correlate with congestion, may help detect CRS in AHF and guide decongestive therapy.[218] Novel AKI indicators' negative predictive power in separating functional serum creatinine variations from actual AKI may be their most important purpose.[15] This distinction at the bedside may influence or guide goal-directed therapy for CRS. However, tubular biomarkers require baseline renal function and may be inaccurate at low GFR. Finally, biomarkers that indicate the AKI-CKD continuum's transition to chronicity may help characterize the acute and chronic CRS and guide therapeutic therapy and prognosis.[15]
Cardiac Biomarkers in Cardiorenal Syndrome
It was reported that in 2017 and 2022, the American College of Cardiology (ACC), American Heart Association (AHA), and Heart Failure Society of America (HFSA)(ACC/AHA/HFSA) guidelines for HF reiterated the Class 1A recommendation for inactive cleavage of pro-BNP and BNP in HF diagnosis/exclusion, prognosis, and severity quantification in chronic and AHF. [219,220] CKD patients had greater baseline BNP levels than comparable individuals with normal renal function due to reduced renal clearance of BNP (especially NT-proBNP), chronic pressure/volume overload, and CKD-associated cardiomyopathy.[221,222] BNP levels are also much higher in CRS patients than in AHF without renal impairment.[223]
Further study is needed to interpret BNP variations in CRS patients receiving angiotensin receptor blocker (ARB)/neprilysin inhibitor medication. In return for biomechanical pressure, the aortic root tract and LV endothelial cells release a tumorigenicity 2 (ST2) suppressor, a decoy protein.[224] ST2 attaches to the IL-33 receptor on cardiac muscle cells and heart satellite cells, causing myocyte malfunction and tissue fibrosis. ST2 levels are a reasonable predictor for HF-related mortality and hospitalizations and are unaffected by renal function.[15]
Cardiac macrophages generate Galetin-3, a β-galactoside-binding lectin that interacts with laminin, synexin, and integrins.[15,220] In 232 NYHA class III or IV HF patients, Lok et al. used eGFR and NT-proBNP to adjust for heart disease severity and renal dysfunction.[225] It was found that galectin-3 blood levels independently predicted CV mortality.[226] Galectin-3 levels that increased by >15% over 3 to 6 months were associated with a significantly higher adjusted risk for all causes of HF hospitalization and mortality in a secondary analysis of the Comparison of Outcomes and Access to Care for Heart Failure (COACH) and Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA) trials.[227] Tang et al. found in a single-center study of chronic HF patients that higher galectin-3 levels were associated with worse renal function and poorer survival and that galectin-3 remained an independent predictor of all-cause mortality in a multivariate analysis of several factors, including eGFR.[224] Furthermore, Lee et al. reported that a hospital-based approach to enhance clinical decision-making and timely follow-up for AHF patients requiring emergency treatment reduced the composite risk of mortality from any cause or CV hospitalization within 30 days after normal care.[228]
Diagnostic and prognostic indicators for acute myocardial damage include high-sensitivity cardiac troponins T and I. When they are raised in acute decompensated HF without myocardial ischemia or coronary artery disease, cardiac troponins have predictive significance and are linked with greater mortality risk.[220] Elevated cardiac troponins rise with diminishing GFR and increase mortality risk.[229] Their increase in low eGFR is sometimes a misleading biomarker in CRS and RCSs.
Imaging Diagnostic Modalities in Cardiorenal Syndromes
Approximately 40% of patients admitted due to AHF have aCRS.[2] The decrease in kidney blood flow caused by increased CVP is a crucial factor, along with a decrease in CO, in the development of AKI in RCS and CRS.[230] Noninvasive imaging techniques are crucial for identifying indicators of venous congestion and reduced forward flow in CRS and RC Patients. These techniques are easily accessible clinical tools that can be used at a patient's bedside. Echocardiography (ECHO) may assist in the diagnosis of congestive conditions by assessing hemodynamic parameters such as central venous pressure (CVP), systolic pulmonary artery (PA) pressure, pulmonary capillary wedge pressure (PCWP)/left atrial pressure, and CO.[231] In addition, additional valuable echocardiographic measures include assessing the longitudinal motion of the lateral and septal walls (E') concerning the velocity of the mitral input (E). The E/E′ ratio was directly correlated with PCWP. An E/E′ ratio greater than 15 indicates a PCWP of 18 mm Hg or higher.[232] Furthermore, ECHO provides predictive information related to CRS phenotypes. Retrospective cohort research showed that aCRS and aRCS had the most significant mortality risk compared with CKD without CRS features. One study found that CRS type 4 patients had a higher survival rate than those with aCRS. Among the patients diagnosed with cCRS, 16% had aCRS, whereas 20% had cRCS acquired aRCS. In addition, 14% of patients with aCRS develop chronic HF or CKD. The incidence of CRS types was shown to be more significant when there was a decrease in LVEF, an increase in PA pressure, and a larger RV diameter, with each factor separately contributing to the association.[233]
Speckle ECHO with strain analysis provides a more comprehensive evaluation of myocardial systolic function in individuals with a normal LVEF. This may provide additional benefits compared to traditional echocardiographic measurement of EF, particularly in cCRS.[234] During a study involving 40 control subjects and 90 patients with CKD, patients with CKD had significantly lower LV longitudinal systolic strain, late diastolic strain rates, and early diastolic strain rates than those in the control group. The differences were significant (P<0.001) for all measurements despite the overall preservation of EF.[235] Global longitudinal strain was a strong indicator of all-cause mortality in CKD.[236]
Renal ultrasonography and analysis of intrarenal venous flow patterns are becoming more critical methods for detecting renal venous congestion and understanding its clinical implications in CRS. Iida et al. investigated the intrarenal venous flow patterns using intrarenal Doppler ultrasonography.[237] These flow patterns were linked to renal artery pressure and significantly correlated with the clinical outcomes. Among the 217 patients with AHF, 54% showed a consistent flow pattern of blood inside the kidneys, which was associated with low right atrial (RA) pressure (estimated to be less than 10 mmHg) and a favorable prognosis (survival rate of over 95% at 1 year). In contrast, approximately 25% of patients with interrupted blood flow inside the kidney, with elevated right atrial pressures (26%) or a single-phase flow pattern (23%), have the worst outcome, with a survival rate of less than 40% after one year. [237] In HF patients, an increase in blood volume inside the blood vessels leads to a notable decrease in blood flow (BF) in the renal veins, even before a considerable rise in pressure within the heart chambers. This decrease in renal venous BF is accompanied by reduced effectiveness of diuretic medications.[238] While renal arterial resistance and renal perfusion indices correlate with CVP, mean arterial pressure, and adequate renal plasma flow, they are unreliable predictors of clinical outcomes in CRS. [237] Renal ultrasonography can determine the chronicity of a disease by examining the kidney size, cortex thickness, echogenicity level, and abnormal corticomedullary ratios. These factors help identify progression from type 1 CRS to a slower developing cCRS phenotype. Additionally, renal ultrasonography can help establish whether AKI or CKD is the principal issue in the CRS clinical presentation.[239]
Cardiac magnetic resonance imaging (MRI) is an established non-invasive technique for evaluating the size and performance of the ventricles and detecting fibrosis. Myocardial fibrosis in type 4 CRS arises from many processes that are not exclusively associated to coronary artery disease. Initial efforts to measure myocardial fibrosis in ESRD using gadolinium-enhanced cardiac MRI revealed a significantly late gadolinium enhancement appearance, which is indicative of coronary artery disease. Additionally, a distinct pattern of fibrosis, unrelated to heart attacks, was observed, suggesting a more widespread presence of fibrotic tissue.[240] Two recent studies have found a solution to the limitations of using gadolinium in progressive CKD due to the risk of nephrogenic systemic fibrosis. These studies have described prolonged native T1 relaxation time and abnormal global longitudinal strain with prevalent HF with preserved ejection fraction (HFpEF), as well as conducting hemodialysis compared to control subjects.[15,241,242] Validation of cardiac MRI without using gadolinium in advanced CKD represents a new opportunity for detecting early LV dysfunction and is a promising prospect for future research on heart structure in CRS.
Addressing fluid overload is the primary focus of therapy to optimize the harmful cycle that occurs in RCS and CRS. Nevertheless, the most effective approach to evaluate fluid status and estimate the ideal weight and suitable decongestion in individuals with DHF or renal illness remains a matter that has not been addressed. This section explains the function of several modalities that may be used in clinical examinations to determine volume status.
Bioimpedance Vector Analysis
Bioimpedance vector analysis (BIVA) is a non-invasive assessment method of bedside volume. It relies on the principle that the body is an electrical circuit with a specific resistance (which represents the opposition to current flow between intracellular and extracellular solutions) and reactance (representing the cells' ability to store energy). BIVA assesses total body water by placing electrodes on the same side of the ankle and wrist and administering a 50-kHz current. The BIVA vector length can be used as an indicator of hydration. Shorter vectors indicate volume overload, whereas longer vectors indicate volume depletion. BIVA has shown encouraging outcomes in differentiating dyspnea resulting from HF from other reasons in patients who arrive at emergency departments.[15,243] BIVA has also been used with BNP to determine the optimal time for AHF discharge, thereby minimizing the occurrence of AKI when high-dose diuretics are administered for HF. Additionally, BIVA has shown promise in predicting the likelihood of rehospitalization and CV fatality in high-risk patients.[15,244] A research had used body composition analysis by bioimpedance to determine a measure of fluid overload. This measure was approved as a decisive factor in managing mortality, regardless of whether the value was low or high.[220]
No definitive intervention or treatment exists for direct management of CRSs. Thus, prevention is the most optimal and effective course of action. CRS development is highly probable in patients with heart or kidney dysfunction. Nonetheless, combating conventional CV disease risk factors, such as HTN and DM, and quitting smoking are among the most effective strategies to confront CRS. Patients with CVD who develop CHF have a negative impact on prognosis.[6,245,246] Adding angiotensin-converting enzyme inhibitors (ACEi) to conventional treatment reduces the incidence of chronic HF decompensation and HF-associated hospitalization in patients with reduced EF and HF. According to several studies, an indirect correlation may exist between the use of RAAS inhibitors and the overdiuresis and increased calcium channel blocker utilization that patients undergoing treatment for DCHF experience a deterioration of renal function.[6,246] Additionally, it is crucial to refrain from administering drugs that induce renal damage directly, such as anti-inflammatory medications and contrast agents, especially in patients who have or are at an increased risk of developing CHF.[6,245,246] Anemia treatment and good nutrition are also effective in preventing CRS. Despite these preventive measures, the risk remains high, particularly in those with renal or cardiac health issues.
Initially, decreased CO or insufficient forward flow was believed to be the main cause of kidney damage. However, the prevailing view today is that venous congestion, rather than a lack of forward flow, is the major cause of renal function deterioration.
No evidence suggests that CRS therapies improve outcomes; treatment focuses on addressing the underlying cause and mitigating complications. Because volume overload affects most CRS patients, fluid removal via ultrafiltration (UF) or diuresis is the treatment keystone for acute CRS. In patients with resistant diseases, alternative therapies like inotropes are reserved.[9] The below therapy approaches are the primary methods used in managing cardiorenal syndrome.[15] CRS therapies can be categorized into three main categories: A) decongestive treatments, B) Neurohormonal Modulation, and C) RAAS inhibition.
Reduction of Body Fluid Content
Diuretic
At present, diuretics remain the preferred medication for first-line treatment of stable individuals with type I CRS. Nevertheless, studies have not shown that diuretics alone improve severe cardiac outcomes.[247] Diuretic treatment aims to decrease visible signs of fluid retention, such as increased jugular venous pressure (JVP) and swelling in the extremities.[220] The study discovered a correlation between enhanced cardiac performance and better renal function in individuals with acute and chronic CRS.[248]
Treatment of ACRS typically entails administering intravenous diuretics to induce aggressive diuresis. For this purpose, loop diuretics, the most potent class of diuretics, are the first-resort medicines. While it is possible to combine loop diuretics with other diuretic classes, their standalone use is neither practical nor advised.[9] Resistance response to standard doses of loop diuretics commonly occurs in patients with acute CRSs. These patients develop resistance to diuretics via a variety of mechanisms.[249] CRS and RCS exhibit aberrant diuretic pharmacokinetics. Except for mineralocorticoid antagonists (spironolactone and eplerenone), all diuretics reach their targets on the endothelium of the renal tubules in an inefficient amount if not when the GFR is incredibly low because of their high protein binding, which makes their filtration impossible. Organic anion transporters are responsible for loop diuretics, carbonic anhydrase inhibitors, and thiazide secretion from the PCT.[9,250] Conversely, the organic cation transporter 2 is responsible for the secretion of epithelial sodium channels that can be inhibited by amiloride and triamterene.[251] Renal dysfunction results in the accumulation of diverse uremic toxins within the body, which vie with diuretics for transportation into PCT via the transporters mentioned above.[252] Strong effects of SNS and RAAS activation increase the tubular sodium uptake, thereby attenuating the diuretic effect.
Patients with a creatinine clearance below 15 mL/min secrete 10% to 20% less loop diuretic into the renal tubules compared to healthy individuals in diuretic dosage.[253] Due to this consequence, diuretic dosage adjustments are necessary during uremia. In AKI, the maximum allowable intravenous infusion furosemide dose is 160-200 mg. This contrasts with patients with preserved renal function for whom the maximum allowable dose is 40–80 mg. Dosage adjustments were necessary when thiazides were administered in combination with loop diuretics. When the GFR is below 20 mL/min, 100-200mg of hydrochlorothiazide is the recommended daily dosage.[253] Dose adjustments for other diuretics in the presence of renal insufficiency remain uncertain; nevertheless, it is advisable to adhere to the upper limit of the usual dose range.[9]
An additional approach to enhancing drug delivery is to administer the prescribed dosage of loop diuretics during renal insufficiency. Continuous infusion abolishes post-diuretic sodium retention and offers a more constant and sustained drug delivery than bolus therapy. A comparison of 308 ADHF patients who received furosemide bolus therapy in the Diuretic Optimization Strategies Evaluation (DOSE) trial assessed the safety and efficacy of the two protocols.[254] Neither group exhibited any discernible variation in net fluid loss or symptom management after 72 hours. Other studies have demonstrated continuous infusion produces more diuresis than bolus equivalent dosage.[255] However, insufficient conclusive evidence supports the maintenance of standard continuous-loop diuretic doses. Combining diuretic therapy with sequential nephron blockade is a crucial therapeutic approach for combating diuretic resistance. Research has demonstrated that urinary output-guided diuretic therapy outperforms conventional diuretic therapy.[254] When high concentrations of single-loop diuretics fail to induce the intended diuretic response, combination diuretic therapy may be subsequent in these therapeutic protocols.
The Acetazolamide in Decompensated Heart Failure with Volume Overload (ADVOR) trial, conducted in 2022, showed that intravenous acetazolamide plus loop diuretics may enhance the process of removing excess fluid in patients with acute HF, resulting in more effective decongestion.[256] Throughout 2023, several planned investigations of ADVOR have been released, delving deeper into the specifics of the initial results. Elevated levels of bicarbonate in the blood, formerly thought to be a result of simple contraction alkalosis, may instead indicate the activation of SNS and RAAS in HF.[257,258] A sub-study of ADVOR researched serum bicarbonate's effect on the effectiveness of acetazolamide since it diminished blood bicarbonate levels while increasing serum chloride levels.[257] Research has revealed that the medicine may effectively alleviate congestion across all bicarbonate levels, with a more pronounced therapeutic effect seen in individuals with greater blood bicarbonate levels. In another study, researchers found that the LV EF does not influence the beneficial benefits of acetazolamide in acute HF.[259] Nevertheless, there was an indication that the slight increase in serum creatinine levels observed when acetazolamide was used for decongestion might be more significant in cases of HF with diminished EF. Two additional studies discovered that the advantages of acetazolamide (increased sodium and water excretion) remain consistent regardless of renal function and that this medication does not cause clinically significant low potassium or sodium levels.[260,261]
Specific clinical scenarios dictate the intended diuretic response. In patients with severe obstruction, it is ideal if the total fluid output exceeds fluid consumption by a minimum of 2-3 L within the initial twenty-four hours. On occasion, intensive care unit patients receive multiple infusions of vital drugs, resulting in a net intake of 1–2 L. The intended urine output in these patients would be even greater than in patients not receiving these infusions.
In the dense part of the Henle loop, loop diuretics obstruct sodium re-absorption. This results in a reduction in renal medullary osmolarity and disruption of the countercurrent exchange mechanism, which inhibits water reabsorption. However, the DCT sodium-chloride cotransporter and sodium-epithelial channel can reabsorb unabsorbed sodium, attenuating the diuretic effect. The combination of loop diuretics with potassium-sparing diuretics or thiazides serves this purpose.
Similarly, carbonic anhydrase inhibitors (e.g., acetazolamide) decrease PCT sodium reabsorption, causing an excessive sodium load in the DCT. DCT's ability is lower than that of PCT; however, a reasonable amount of sodium reabsorption occurs in DCT. Therefore, loop diuretics and acetazolamide may exert synergistic diuresis when combined.
The most frequently prescribed combination consists of a thiazide and a loop diuretic despite the absence of extensive placebo-controlled trials.[262,263] Metolazone, a thiazide-like diuretic, is frequently prescribed owing to its accessibility and affordability.[264] Further evidence suggests that PCT sodium reabsorption inhibition by metolazone is a property that potentially has a substantial synergistic effect. Chlorothiazide exhibits a more rapid onset of action than metolazone and can be administered intravenously. Nonetheless, research has been unable to identify any advantage over metolazone.[265,266] Concurrent loop diuretics and acetazolamide administration may result in a reduced propensity for metabolic alkalosis, a possible adverse effect of both loop diuretics and thiazides. A recent study demonstrated that adding bumetanide to acetazolamide substantially increased natriuresis despite the paucity of data.[267–269] The addition of large concentrations of spironolactone to standard therapy did not produce any statistically significant alteration in total urine output and N-terminal pro-B-type natriuretic peptide level, as determined in the Aldosterone Targeted Neurohormonal Combined With Natriuresis Therapy in Heart Failure (ATHENA-HF) trial.[270]
Ultrafiltration
Iso-osmolar fluid UF is UF at a constant rate through a venovenous extracorporeal circuit known as UF.[271,272] Contemporary UF systems are characterized by increased portability, peripheral venous access compatibility, and minimal nursing oversight.[272,273] Despite the potential appeal of UF as a substitute for diuresis in acute HF cases, research findings have been equivocal. The Cardio-renal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) studied 188 acute CRS patients to compare UF and diuresis.[254,274] When executed by an algorithm, Diuresis was determined to be more favorable than UF concerning a bivariate endpoint involving weight change and Scr level change at 96hs. In contrast, it is believed that cystatin C level provides a more precise renal function assessment, and there was no noticeable difference in the CysC level change from baseline between the two treatment groups. Furthermore, intravascular depletion may have resulted from an excessive UF rate of 200 mL/h.
Although the ideal fluid removal rate remains unknown, it is mandatory to customize and modify it according to the patient's hemodynamic status, renal function, and fluid volume. The degree of fluid excess and the expected rate of plasma replenishment from the interstitial fluid should determine the initial rate.[37] Malnourished patients, for instance, might exhibit diminished plasma replenishment upon UF because of their hypovolemic state induced by low oncotic pressure. A perturbation in the intricate equilibrium between the plasma replenishment rates and UF can produce contraction in the intravascular volume.[9] UF is used in patients with diuretic resistance to ADH and compromised renal function. However, it is not a successful treatment for CRS. Further research is needed to determine if UF might benefit patients with frequent rehospitalizations for ADHF and functional diuretic resistance. This research will assess whether meaningful and clinically significant effects can be obtained in these high-risk individuals.[275,276]
Different modules to remove fluids by using extracorporeal devices are in practice. The available choices for extracorporeal UF include slow continuous ultrafiltration (SCUF), isolated UF using a conventional dialysis machine, and continuous renal replacement therapy (CRRT), particularly in the continuous venovenous hemofiltration (CVVH) mode. SCUF can be conducted continuously with a blood flow rate of 50 to 100 mL/min. It can be configured to accomplish volume control by providing a UF rate between 100 and 300 mL/h. Isolated UF is a process that utilizes conventional dialysis equipment to remove fluid from the body without dialysis. Each session of isolated UF typically lasts for 4–6 hours. The rate of fluid loss is often higher, which may lead to a higher occurrence of hemodynamic problems. SCUF or isolated UF is unsuitable for patients requiring solute clearance because these methods are designed only for fluid removal.
UF techniques such as SCUF and CVVH are similar. However, in CVVH, the electrolyte concentration in the replacement solution can be adjusted to control the body's electrolyte balance, particularly that of sodium. This allows for the removal of sodium to be separated from the removal of water, resulting in the achievement of normal fluid and electrolyte levels, particularly in the extracellular compartment.[277,278]
In brief, while UF can act as a beneficial substitute for diuretics in instances of diuretic resistance, its efficacy as a principal decongestive therapy is questionable, given the available evidence. Therefore, extensive randomized studies are required to confirm this hypothesis.
Tolvaptan
Activation of the RAAS pathway in HF leads to elevated ADH levels. This has negative consequences on cardiac function, causing a decline in heart performance. Additionally, it results in peripheral vasoconstriction and an increase in afterload via activation of vasopressin V1 receptors (V1a). Moreover, stimulating the vasopressin V2 receptor causes water retention and a rise in preload.[279,280] Tolvaptan has shown a beneficial effect on HF by decreasing the fluid load. It also aids in weight loss, promotes increased urine production, and corrects blood sodium levels without affecting serum renal function parameters or electrolyte levels. This is achieved via its effect on the neurohormonal mechanism in CRS development.[279,281]
The Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study with Tolvaptan (EVEREST) included 4133 patients. This revealed that tolvaptan successfully decreased the volume overload and provided symptomatic relief. However, it did not demonstrate any improvement in mortality or morbidity.[282] It has a significant safety profile and effectively alleviates congestion without adversely affecting kidney function in individuals with excessive fluid volume and compromised heart and kidney function.[283] Tolvaptan may relieve congestion and help avoid or reduce renal complications by maintaining kidney perfusion and preventing intravascular volume depletion.[284]
Improving Cardiac Output
Inotropes
Cardiogenic shock commonly necessitates the administration of inotropes such as dobutamine and milrinone to preserve organ perfusion. Utilizing inotropes in acute CRS is also physiologic, particularly when the abovementioned approaches fail to surmount diuretic resistance.[14,285] Inotropes alleviate systemic congestion by increasing kidney BF, enhancing right heart output, and increasing CO. These hemodynamic responses have the potential to enhance diuretic effects and renal perfusion. However, the absence of clinical evidence supports this claim. Intermittent mechanical and inotropic support for circulation should only be utilized as a last intervention.
The Renal Optimization Strategies Evaluation (ROSE) study involved 360 patients with AKI and transient HF. The study concluded that a modest dose of dopamine (2 g/kg/min) added to diuretics had no significant effect on CysC levels, 72-hour renal function, or total urine output.[286,287] However, AKI was not identified throughout the study, and it is plausible that some patients were hospitalized with elevated renal function.
Beta Blocker and Renin-Angiotensin System Effect Inhibition
Research has shown that beta-blockers positively impact EF in HF, reduce symptoms, and extend life expectancy. Nevertheless, evidence of their efficacy in CKD patients is scarce.[220] Furthermore, inotropes can improve CRS by augmenting CO and reducing venous congestion. Despite some progress, the effectiveness of inotropes and vasodilators for acute CRS treatment has not yet been shown.[15] Although inotropes have been shown to enhance the CO and EAFV in systolic HF, their adverse effects, including arrhythmias and cardiac ischemia, have limited their usefulness. This was demonstrated in the "Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF)" study, which showed increased mortality and poorer outcomes in the milrinone arm.[288] Milrinone enhances cardiac contractility, inotropy, cardiac relaxation, or lusitropy, and promotes vasodilation.[289]
A phosphodiesterase inhibitor, specifically levosimendan, has been investigated in CRS. In one study, levosimendan enhanced GFR when contrasted with dobutamine.[290] Nevertheless, another investigation of levosimendan and dobutamine failed to demonstrate any advantage.[291] As yet, the function of inotropic agents in CRS remains uncertain.
Moreover, RAAS suppression, specifically via the use of ACEis and ARBs, is a commonly prescribed treatment for HF with a lower EF. However, it is essential to note that this medication does not often improve renal function.[15,275] The long-term effectiveness of completely suppressing the RAAS with an ACEI or ARB is constrained by the occurrence of aldosterone escape, which leads to elevated levels of blood aldosterone. When mineralocorticoid receptor antagonists are used with an ACEI/ARB, they may further decrease the RAAS, potentially leading to long-term advantages for the heart and kidneys.[280,292]
As type 2 DM, HF, and CKD frequently coexist and share a common pathophysiological background, adopting a comprehensive therapeutic approach targeting comorbidities may synergistically affect patient health, significantly improving outcomes. Large-scale clinical trials consistently show that novel glucose-lowering drugs, such as SGLT2i and glucagon-like peptide 1 receptor agonists (GLP-1 RA), improve glycemic control and reduce important CV and renal endpoints in type 2 DM patients.[93,293,294]
Sodium-glucose cotransporter-2 (SGLT2) inhibitors
SGLT2 inhibitors, sustained by their natriuretic and osmotic properties, are believed to offer significant cardiorenal protection.[295] Recently, there has been an increasing interest in studying the impact of medicines that target PCT, such as SGLT-2 inhibitors, on acute HF and CRS. This change in focus moves away from the conventional emphasis on enhancing salt reabsorption in the distal convoluted tubule because SGLT2 inhibitors can be used in patients with eGFR ≥ 25 ml/min/1.73 m2. In 2023, there was a remarkable continuation of the success of SGLT-2i, as shown by the publication of several papers. The Empagliflozin in Patients with Chronic Kidney Disease (EMPA-KIDNEY) trial reported that empagliflozin has renal and cardiac protection.[296] Within this global randomized controlled trial, including a cohort of over 6,600 individuals diagnosed with CKD (with an eGFR ranging from 20 to 90 mL/min/1.73 m2), the administration of empagliflozin resulted in a substantial reduction of 28% in the likelihood of both renal disease progression and cardiovascular death. The experiment was prematurely terminated after 2 years owing to the effectiveness of the intervention arm. Empagliflozin was effective in diabetic and non-diabetic individuals, although to a lesser extent in the latter group. The EMPA-KIDNEY study provided additional data supporting the use of gliflozins in CKD treatment. It is worth mentioning that the subgroup analysis indicated that the positive benefits of empagliflozin were less apparent in patients who did not receive RAAS inhibition.
The purpose of Dapagliflozin Evaluation to Improve the Lives of Patients with Preserved Ejection Fraction Heart Failure (DELIVER) study was to evaluate the effects of dapagliflozin on preserved EF patients to improve their quality of life. The DELIVER study demonstrated that dapagliflozin could potentially decrease the likelihood of worsening HF or CV mortality in over 6,200 individuals with HF and a modest decline or preserved EF (with a median EF of 54%).[297] The increased Scr levels were previously seen as a possible safety issue because of the SGLT-2i natriuretic and osmotic diuretic actions. A secondary assessment of the DELIVER study investigated the consequences of an initial decrease in the eGFR. eGFR reduction was common with dapagliflozin, with 40% of the patients experiencing a drop of > 10%. However, this initial decline was not linked to an increased CV or risk of kidney events.[298] It is worth mentioning that earlier reports have shown that the first decrease in eGFR does not negatively affect the outcomes in patients with HF and decreased EF.[298]
Furthermore, the effects of gliflozins on the volume status and CRS outcomes were examined in 2023. Within the EMPA-KIDNEY trial, a substudy was conducted to assess the influence of empagliflozin on fluid overload, specifically the amount of excess extracellular water, in 660 patients with CKD. Bioimpedance measurements were performed in this evaluation. A study discovered that empagliflozin was linked to a consistent decrease in fluid overload.[299] Furthermore, a multicenter, open-label, randomized controlled trial was conducted to evaluate the diuretic efficacy of dapagliflozin with metolazone in acute HF patients and diuretic resistance.[300] They found no considerable difference in weight loss between the two groups. However, patients who were administered dapagliflozin received a higher total furosemide dose and had a smaller decrease in their blood salt levels. Recent research indicates that gliflozins may have further positive effects on fluid and electrolyte balance, which has not yet been thoroughly investigated in CKD and HF. A preliminary randomized controlled trial indicated that empagliflozin might be a potentially effective therapy for hyponatremia associated with chronic inappropriate secretion of ADH syndrome. Furthermore, it may also potentially enhance neurocognitive performance in these individuals.[301] The efficacy of SGLT-2i persists, and these drugs have become a well-established treatment for CKD, HF, and DM. However, ongoing research suggests that these agents may also be beneficial in other conditions, such as acute HF and hyponatremia.
The Empagliflozin, Cardiovascular Outcome (EMPA-REG OUTCOME) trial investigated the impact of empagliflozin on CV morbidity and mortality in 7020 patients with DM type 2 who were taken the standard of care.[302] The study demonstrated that 10.5% of individuals who took empagliflozin had composite CV events (including CV mortality, nonfatal stroke, and myocardial infarction), whereas 12.5% of patients who were administered a placebo experienced the same events. In comparison, the combined renal outcome (which includes a doubling of Scr level, the need for renal replacement therapy, and renal mortality) was found to be 1.7% in the treatment group. In contrast, the rate was 3.1% in the placebo group. The research further showed that the medication led to a noteworthy decrease in CV risk variables such as body waist circumference, weight, uric acid levels, and both diastolic and systolic blood pressure, without any increase in heart rate.[302]
Neal et al. conducted the Canagliflozin Cardiovascular Assessment Study (CANVAS), which consisted of two related trials. These trials aimed to investigate the effectiveness of canagliflozin in improving heart and kidney outcomes. The results showed that the composite CV outcome occurred in 26.9 out of 1000 patients who received canagliflozin, compared to 31.5 out of 1000 patients who received a placebo. Additionally, renal outcomes were observed in 5.5 out of 1000 patients who received canagliflozin compared to 9 out of 1000 patients who received a placebo. The research ascribed the advantages to enhanced regulation of blood sugar levels, drop in blood pressure, lowered intraglomerular pressure, decreased albuminuria, and improved excessive fluid volume.[303]
In 17,160 patients, Wiviott et al. demonstrated that 8.8% of patients who received dapagliflozin experienced a favorable CV outcome compared to 9.4% of patients who received a placebo. Additionally, 1.5% of patients on the drug experienced renal outcomes compared to 2.8% of patients on the placebo.[293] The Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) trial evaluated the effects of canagliflozin on patients with established nephropathy and involved 4401 participants. The trial found that the composite CV outcome was 9.9% in patients who received canagliflozin compared to 12.2% in patients who received a placebo. Additionally, the trial showed that renal outcomes were 11.1% in canagliflozin patients, whereas placebo patients had a renal outcome rate of 15.4%. The experiment ascribed the results to RAAS inhibition and decreased pressure inside the glomerulus.[304]
The potential advantages of SGLT2 inhibitors (SGLT2i) were discovered to result from lower stiffness in both the systemic and renal arteries, reductions in hyperglycemia, hyperlipidemia, and the expression of inflammatory markers. The upregulation of sodium-glucose co-transporter-1 inhibitors (SGLT1) receptors in cardiomyocytes might be a promising therapeutic target for protecting the heart.[305] The effects of decreasing blood pressure and reducing intravascular volume due to osmotic diuresis are believed to occur because sodium reabsorption, which is co-transported with glucose, is inhibited.[306] SGLT2i also enhances the secretion of adenosine by increasing the transport of sodium to the macula densa. This process activates tubulo-glomerular feedback, causing the return of the afferent arteries to their size as before vasodilation and decreasing the pressure inside the glomerulus.[307] These pharmacological groups decrease urate levels by increasing the rate at which urate is eliminated via the kidneys by suppressing the activity of Glucose Transporter 9b (GLUT9b).
Caloric control and adaptive ketogenesis contribute to the preservation of the CV system. Elevated glucose excretion leads to a transition towards fat utilization, enhanced glucagon synthesis and release, and increased peripheral insulin levels. This, in turn, triggers the release of free fatty acids, improves ketogenesis, and improves cardiac metabolism. The change in metabolism reduces the amount of oxygen used by the kidneys, which helps relieve the stress caused by lack of oxygen and slows down the advancement of kidney damage.[306]
In addition, SGLT2i lowers the fat content in the epicardia. This reduction in epicardiac fat may decrease harmful stimuli such as leptins and RAAS components, which are implicated in CV inflammation and fibrosis. The renoprotective SGLT2i effects include diuresis and natriuresis.[308] The drug's impact on natriuresis is partially caused by the breakdown of the functional connection between SGLT2 and sodium proton exchanger 3 (NHE3).[309]
Interestingly, it has been reported that SGL2i prevents CRS through different mechanisms. These mechanisms include reducing blood volume, altering cardiac substrate preference, weight loss, decreasing atrial stiffness, declining the adipocytes account and activity, decreasing systolic blood pressure, inhibiting inflammatory reactions, controlling blood sugar, diverting the catabolic substrate of cardiac muscles to fat (inducing ketogenesis) and improving hemoglobin levels.[280] Figure 4 illustrates the mechanisms of SGL2i for CRS prevention.
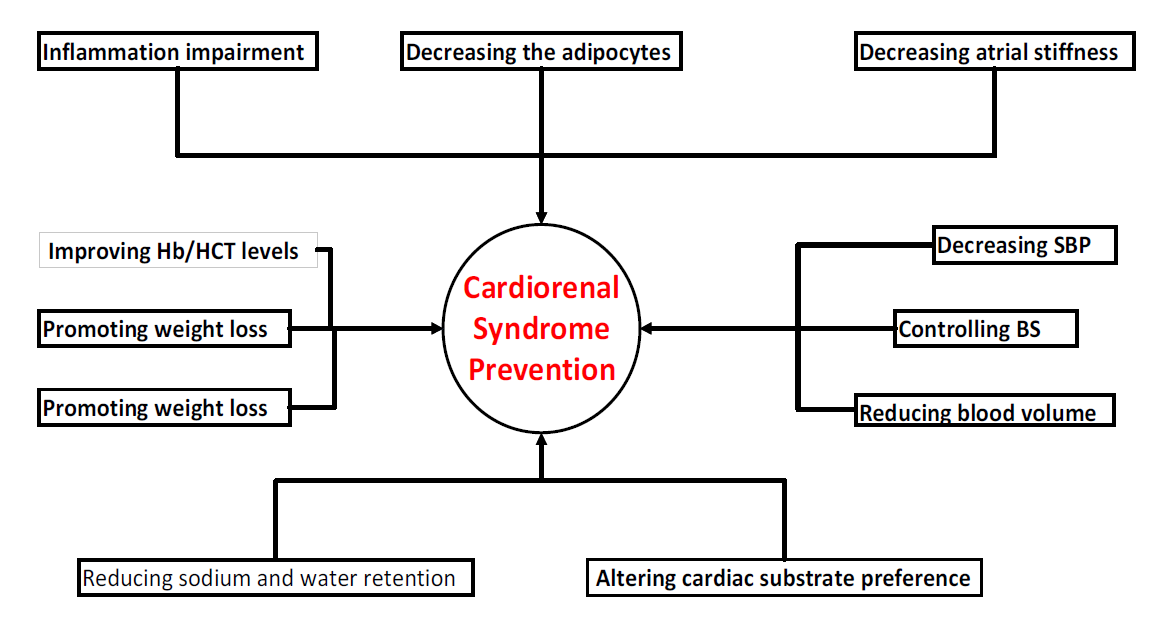
Figure 4. Mechanism of SGL2 inhibitors for CRS prevention.
Abbreviations: Systolic blood pressure (SBP), hemoglobin (Hb), hematocrit (HCT), and blood sugar (BS).
Glucagon-like Peptide 1 Receptor Agonists
GLP-1 RA may exert protective effects through anti-atherogenic and immune-modulating mechanisms. [310,311] It is used in type 2 DM to regulate blood sugar levels and reduce weight in both diabetic and nondiabetic overweight or obese individuals. [294,312] GLP-1 RA therapy also controls hypertension, [294,313] reduces atherosclerosis,[312] and decreases the risk of CV death, [312,313] and stroke.[294,310] Furthermore, GLP-1 RAs improve kidney function by reducing proteinuria,[294] which may have implications for the prevention and treatment of chronic kidney disease.[312] However, further research is required to confirm the effectiveness of GLP-1 RAs in these areas.
The new bi-receptor agonist (tirzepatide) stimulates two pathways that depend on incretin, which are both simultaneously activated.[314] It operates by imitating the actions of GLP-1 and GIP hormones that are naturally produced by the intestine following a meal, leading to insulin secretion and appetite reduction. By slowing down the rate at which the stomach empties and interacts with brain regions containing GLP-1 receptors, tirzepatide signals a feeling of fullness. The synergistic effect of this duality has a positive impact on both glycemic and weight control, resulting in improved metabolic outcomes when compared to selective GLP-1 RA.[315]
This medication has recently been approved for weight loss. [314,315] It has been reported to aid in the control of blood sugar levels, [314,315] may also contribute to the reduction of CV risk factors,[314] and potentially improve proteinuria while preventing CKD progression in type 2 DM patients. However, further research is required to confirm these effects of tirzepatide.
Cardiac Resynchronization Therapy
Cardiac resynchronization is a therapeutic intervention aimed at reinstating the regular rhythm and timing of the heartbeat. A dual-chamber pacemaker (in both the right atrium and ventricle) can synchronize the timing of the atria and ventricles of the heart. Cardiac resynchronization therapy was used to treat HF. Additionally, it has enhanced renal function via improved CO, mean arterial pressure elevation, and reduction of CVP.[316]
A retrospective cohort study of 260 patients revealed that individuals with CKD and CHF improved renal responsiveness after cardiac resynchronization therapy. In addition, there was a notable decrease in death, transplant, and LV assist device use within five years. This improvement was observed in stage 4 CKD patients. The reduction in these events was attributed to an enhancement in LV EF, resulting in improved blood flow and reduced venous congestion.[317] Cardiac resynchronization therapy decreases sympathetic nerve activity, reducing adrenergic tone. This subsequently leads to a decrease in RAAS activity over time, which may explain the improvement in renal function. Further, it was discovered that cardiac resynchronization therapy improves survival in intermediate CKD and HF in CRS.[318]
Nevertheless, a study that included 482 patients demonstrated a greater risk of renal dysfunction after the implementation of a dual pacemaker. This condition is often associated with worse survival outcomes than those in patients with normal kidney function. Additionally, there was an elevated death rate seen in CKD and increased anemia incidence in CKD.[319] Another study revealed that approximately 33% of patients with CRS improved kidney function following cardiac resynchronization therapy. This lack of improvement may be attributed to the presence of intrinsic renal disease, which is linked to severe HF. The authors ascribed the results to systemic alterations that result in cardiac remodeling and deterioration of glomerular filtration, thereby reducing the effectiveness of the resynchronization treatment.[284]
Device Therapeutic Options in Cardiorenal Syndrome
Circulatory devices used to treat aCRS may be classified into two main groups. These devices aim to restore the proper pressure gradient for good renal perfusion by addressing the hemodynamic equation from one side or the other.[320,321] Pusher devices enhance renal arterial perfusion (renal preload) without relying on vasopressor/inotrope administration. In contrast, puller devices aim to alleviate renal venous congestion (renal afterload). The currently available devices are experimental and have not yet been licensed in healthcare centers.[322] This is because they must fulfill the complete need for clinical evidence in patients with CRS. However, exploring using "pusher" and "puller" tactics is theoretically appealing, alone or in combination. Additionally, it would be intriguing to examine the integration of device-based treatments with diverse mechanisms in the future treatment of CRS. See Nathan and Basir (2023) for more information about the devices. [322] The therapy options for RCS are summarized in Table 2.
Table 2. Summary of therapeutic options for cardiorenal syndrome.
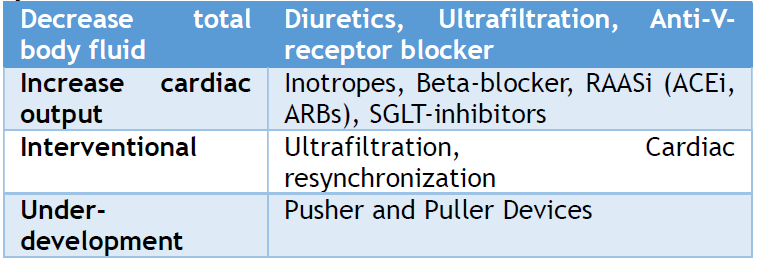
Abbreviations: Renin-angiotensin-receptor-inhibitor (RAASi), Angiotensin inhibitors (ACEi), Angiotensin receptor blocker (ARB), Sodium-glucose Cotransporter-2 (SGLT2) Inhibitors
In severe cases, CRS patients require hospital admission, and the above therapy strategies should be individualized according to each patient's needs. Most, if not all, patients with severe CRS require intensive care unit admission. Despite all the efforts available, the survival rate remains low.
To achieve better outcomes, a multidisciplinary strategy is necessary to manage CRS and RCS in hospitals and even on an outpatient basis. This entails the collaboration of pharmacists, physicians, and other medical specialists, including physician assistants and specialty-trained nurses from various disciplines, to attain the most favorable patient outcomes. Improving the affected patients' outcomes necessitates diligent monitoring of the patient’s symptoms, weight, and laboratory results after discharge from the hospital. An inter-professional team of clinical providers, physician assistants, nurses, and pharmacists was required to attain optimal outcomes.
The pharmacist is responsible for educating patients regarding the importance of administering medications promptly and avoiding those that may have adverse effects on the disease. Effective communication between pharmacists and therapy providers is critical for preventing the initiation of potentially harmful medications that may lead to adverse effects on the patient. Regular monitoring by a specialized nurse and physician assistant is essential to verify that the patient maintains an appropriate weight and complies with prescribed dietary and activity restrictions and medications. Collaboration between the primary clinical provider, kidney and cardiac physicians, and the patient is vital for optimal CRS management and achieving the most favorable outcome. Another critical factor affecting the outcomes of CRS patients is baseline renal function in all types of CRS.
A low baseline GFR in HF is related to poor prognosis. The prognostic significance of WRF depends on its cause. A study of the PROTECT trial found a variety of renal function trajectories following acute HF hospitalization.[323] The most common Scr trajectories in hospitals were temporary (19%), persistent (17.6%), and declining (14.5%). After multivariable correction, no change was significantly related to better or worsening outcomes, casting doubt on the prognostic importance of renal function changes during acute HF.
HF patients may have a 30-60% drop in baseline GFR (< 60 mL/min/1.73 m2). [2,3] This is clinically significant because the GFR at presentation predicts mortality in acute and chronic HF. [247,286,324,325] A meta-analysis of 16 studies comprised over 80,000 HF patients who were categorized into three kidney function groups: normal (eGFR ≥ 90 mL/min), mild (eGFR 53-89 mL/min, Scr > 88.4 µmol/L, or S CysC > 1.03 to 1.55 mg/dL), and moderate to severe (eGFR < 53 mL/min, Scr ≥133 µmol/L). At one year or longer, 24% of patients with normal eGFRs died. The proportions of patients with modest and moderate-to-severe eGFR decreases were 38% and 51%, respectively. Every 10 mL/min fall in eGFR increased the mortality rate by 15%. Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity (CHARM) trial, which comprised 2680 chronic HF patients, found comparable effects during a median follow-up period of > three years.[37] Overall mortality increased significantly when the initial eGFR was less than 75 mL/min/1.73 m2. The adjusted hazard ratio (HR) increased from 1.20 to 2.92 when the eGFR decreased below 45 mL/min/1.73 m2. LVEF did not affect this effect, although all-cause mortality increased when LVEF decreased to < 45%. Another study indicated that 4917 continuous-flow LVAD patients with worse renal function before implantation had a shorter survival rate. An eGFR of < 30 mL/min patients had a 20% poorer two-year survival rate than those with ≥ 60.[247] Survival decreased the most in the first three months after LVAD placement.
In some patient groups, a drop or improvement in GFR increases mortality risk, although the source of a decline affects its prognostic relevance.[2,163,326] Hospitalized patients with HF contributed the most to the GFR change-outcome data. An analysis of 3,570,865 US veterans with an eGFR of 60 mL/min/1.73 m2, of which 156,743 were HF patients, had WRF.[327] Compared with 14.5/1000 patient-years for those without HF, the incidence rate of CKD was 69.0/1000 HF. A precipitous drop in eGFR was reported in 22% of patients with HF compared to 8.5% of non-HF patients. Patients with HF showed a fast eGFR drop and doubled incident CKD risk, a composite of mortality and CKD. A meta-analysis of eight trials, including nearly 18,000 HF patients, shows that impaired renal function was associated with increased death rates.[163] Five trials included hospitalized patients, and three involved outpatients. Patients with an Scr increase ≥ 27 µmol/L had a 26% decline in renal function. Patients with renal function decline had a higher all-cause death risk (43% vs. 36%) than those with Scr unaltered or < 0.2 mg/dL (18 µmol/L). The outcomes were the same in the non-hospitalized and hospitalized patients. The risk of death increases with a decline in renal function. A GFR increase of less than 5 to 10 mL/min/1.73 m2 was associated with a slight rise in mortality risk. Death risk increased when eGFR dropped more than 15 mL/min/1.73 m2 or Scr mounted > 0.5 mg/dL.
Other studies have suggested that patients with increasing or decreasing renal function may have poorer outcomes. Variable renal function in unwell individuals may be linked to a worse prognosis than constant renal function. An examination of 401 ESCAPE trial participants found that ADHF patients whose estimated GFR increased or fell had similar outcomes.[328] Patients whose GFR improved had a lower cardiac index, required inotropes and vasodilators and had a higher all-cause death rate than those with a steady GFR. Another observational study of 903 HF patients found survival worse for those whose GFR improved during hospitalization than those whose renal function remained unchanged.[325] This observation primarily affected the patients with persistent renal failure after discharge. In summary, baseline kidney function is a determining factor for the prognosis of CRS and RCS, particularly RCS.
Cardiorenal syndrome frequently refers to the combined dysfunction of the kidney and heart, which triggers a series of feedback mechanisms that damage both organs. Both syndromes have dynamic, complex, and multifactorial pathophysiology, challenging diagnosis and treatment. A deeper comprehension of the underlying disease mechanisms is essential for the advancement of targeted pharmacological and non-pharmacological interventions to effectively manage this syndrome..
Syndrome treatment is complex due to the complicated heart-kidney relationship. Preventive strategies to alter risk factors, pharmaceutical therapies, hemodynamic support, and collaborative teamwork are essential for achieving good outcomes and prevention. Finally, additional research into innovative technologies and therapies will improve the treatment standards for these syndromes so that healthcare providers may better manage and treat this complicated condition. Further research is needed to investigate the unresolved pathophysiological mechanisms and interventions.
In the subsequent parts of this review, we will describe each type of syndrome individually, addressing all relevant aspects and providing updates..
AUTHORS' CONTRIBUTION
Each author has made significant contributions to the current work, whether in one or more areas, including conception, study design, conduct, data collecting, analysis, and interpretation. They also provided their final approval for the text to be published after taking part in its drafting, revision, or critical assessment..
FUNDING
None
CONFLICT OF INTEREST
None
References
- Ronco C, McCullough PA, Anker SD, Anand I, Aspromonte N, Bagshaw SM, et al. Cardiorenal syndromes: an executive summary from the consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol. 2010;165:54-67.
- Forman DE, Butler J, Wang Y, Abraham WT, O'Connor CM, Gottlieb SS, et al. Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J Am Coll Cardiol. 2004;43:61-7.
- Heywood JT. The cardiorenal syndrome: lessons from the ADHERE database and treatment options. Heart Fail Rev. 2004;9:195-201.
- Goffredo G, Barone R, Di Terlizzi V, Correale M, Brunetti ND, Iacoviello M. Biomarkers in cardiorenal syndrome. J Clin Med. 2021;10:3433.
- House AA, Anand I, Bellomo R, Cruz D, Bobek I, Anker SD, Aspromonte N, et al. Definition and classification of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant. 2010;25:1416-20.
- Ajibowo AO, Okobi OE, Emore E, Soladoye E, Sike CG, Odoma VA, et al. Cardiorenal Syndrome: A Literature Review. Cureus. 2023;15(7): e41252.
- Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527-39.
- Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53:582-8.
- Thind GS, Loehrke M, Wilt JL. Acute cardiorenal syndrome: Mechanisms and clinical implications. Cleve Clin J Med. 2018;85(3):231-9.
- Habas El, Akbar R, Khan F, Elzouki AN. Reno-Cardiac Syndrome (RCS): A Clinical Review. IJSR. 2021;10(6): 1705-1712.
- Kumar U, Wettersten N, Garimella PS. Cardiorenal Syndrome: Pathophysiology. Cardiol Clin. 2019;37(3):251-5.
- Zhang Y, Jiang Y, Yang W, Shen L, He B. Chronic Secondary Cardiorenal Syndrome: The Sixth Innovative Subtype. Front Cardiovasc Med. 2021;8639959.
- Chang CH, Lin CY, Tian YC, Jenq CC, Chang MY, Chen YC, et al. Acute kidney injury classification: comparison of AKIN and RIFLE criteria. Shock. 2010;33(3):247-52.
- Hatamizadeh P, Fonarow GC, Budoff MJ, Darabian S, Kovesdy CP, et al. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol. 2013;9(2):99-111.
- Rangaswami J, Bhalla V, Blair JEA, Chang TI, Costa S, Lentine KL, et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation. 2019;139(16):e840-e878.
- Heywood JT, Fonarow GC, Costanzo MR, Mathur VS, Wigneswaran JR, Wynne J. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. 2007;13(6):422-30.
- Hakahama H, Kitakaze M. Pathophysiology of cardiorenal syndrome in patients with heart failure: potential therapeutic targets. Am J Physiol Heart Circ Physiol. 2017 ;313(4):715-21.
- Gottlieb SS, Abraham W, Butler J, Forman DE, Loh E, Massie BM, et al. The prognostic importance of different definitions of worsening renal function in congestive heart failure. J Card Fail. 2002;8(3):136-41.
- Goldberg A, Hammerman H, Petcherski S, Zdorovyak A, Yalonetsky S, Kapeliovich M, et al. Inhospital and 1-year mortality of patients who develop worsening renal function following acute ST-elevation myocardial infarction. Am Heart J. 2005;150(2):330-7.
- Newsome BB, Warnock DG, McClellan WM, Herzog CA, Kiefe CI, Eggers PW, et al. Long-term risk of mortality and end-stage renal disease among the elderly after small increases in serum creatinine level during hospitalization for acute myocardial infarction. Arch Intern Med. 2008;168(6):609.
- Parikh CR, Coca SG, Wang Y, Masoudi FA, Krumholz HM. Long-term prognosis of acute kidney injury after acute myocardial infarction. Arch Intern Med. 2008;168(9):987-95.
- Jose P, Skali H, Anavekar N, Tomson C, Krumholz HM, Rouleau JL, Moye L, et al. Increase in creatinine and cardiovascular risk in patients with systolic dysfunction after myocardial infarction. J Am Soc Nephrol. 2006;17(10):2886-91.
- Nohria A, Hasselblad V, Stebbins A, Pauly DF, Fonarow GC, Shah M, et al. Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol. 2008;51(13):1268-74.
- Krumholz HM, Chen YT, Vaccarino V, Wang Y, Radford MJ, Bradford WD, Horwitz RI. Correlates and impact on outcomes of worsening renal function in patients > or =65 years of age with heart failure. Am J Cardiol. 2000;85(9):1110-3.
- Smith GL, Vaccarino V, Kosiborod M, Lichtman JH, Cheng S, Watnick SG, et al. Worsening renal function: what is a clinically meaningful change in creatinine during hospitalization with heart failure? J Card Fail. 2003;9(1):13-25.
- Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53(7):589-596.
- Ahmed A, Rich MW, Sanders PW, Perry GJ, Bakris GL, Zile MR, et al. Chronic kidney disease associated mortality in diastolic versus systolic heart failure: a propensity matched study. Am J Cardiol. 2007;99(3):393-8.
- Dimopoulos K, Diller GP, Koltsida E, Pijuan-Domenech A, Papadopoulou SA, Babu-Narayan SV, et al. Prevalence, predictors, and prognostic value of renal dysfunction in adults with congenital heart disease. Circulation. 2008;117(18):2320-8.
- Kellum JA, Levin N, Bouman C, Lameire N. Developing a consensus classification system for acute renal failure. Curr Opin Crit Care. 2002;8:509–14.
- Burns KE, Chu MW, Novick RJ, Fox SA, Gallo K, Martin CM, et al. Perioperative N-acetylcysteine to prevent renal dysfunction in high-risk patients undergoing cabg surgery: a randomized controlled trial. JAMA. 2005;294(3):342-50.
- Leacche M, Rawn JD, Mihaljevic T, Lin J, Karavas AN, Paul S, Byrne JG. Outcomes in patients with normal serum creatinine and with artificial renal support for acute renal failure developing after coronary artery bypass grafting. Am J Cardiol. 2004;93(3):353-6.
- Bove T, Calabrò MG, Landoni G, Aletti G, Marino G, Crescenzi G, Rosica C, et al. The incidence and risk of acute renal failure after cardiac surgery. J Cardiothorac Vasc Anesth. 2004;18(4):442-5.
- Hoste EA, Cruz DN, Davenport A, Mehta RL, Piccinni P, Tetta C, et al. The epidemiology of cardiac surgery-associated acute kidney injury. Int J Artif Organs. 2008;31(2):158-65.
- Cheung AK, Sarnak MJ, Yan G, Berkoben M, Heyka R, Kaufman A, et al. Cardiac diseases in maintenance hemodialysis patients: results of the HEMO Study. Kidney Int. 2004;65(6):2380-9.
- Dries DL, Exner DV, Domanski MJ, Greenberg B, Stevenson LW. The prognostic implications of renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35(3):681-9.
- Foley RN, Murray AM, Li S, Herzog CA, McBean AM, Eggers PW, Collins AJ. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States Medicare population, 1998 to 1999. J Am Soc Nephrol. 2005;16(2):489-95.
- Hillege HL, Nitsch D, Pfeffer MA, Swedberg K, McMurray JJ, et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation. 2006 ;113(5):671-8.
- McCullough PA, Jurkovitz CT, Pergola PE, McGill JB, Brown WW, et al. Independent components of chronic kidney disease as a cardiovascular risk state: results from the Kidney Early Evaluation Program (KEEP). Arch Intern Med. 2007 Jun 11;167(11):1122-9.
- Herzog CA, Ma JZ, Collins AJ. Poor long-term survival after acute myocardial infarction among patients on long-term dialysis. N Engl J Med. 1998;339(12):799-805.
- Johnson DW, Craven AM, Isbel NM. Modification of cardiovascular risk in hemodialysis patients: an evidence-based review. Hemodial Int. 2007;11(1):1-14.
- Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351(13):1296-305.
- McCullough PA, Li S, Jurkovitz CT, Stevens LA, Wang C, Collins AJ, et al. CKD and cardiovascular disease in screened high-risk volunteer and general populations: the Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis. 2008 Apr;51(4 Suppl 2):S38-45.
- McCullough PA, Li S, Jurkovitz CT, Stevens L, Collins AJ, Chen SC, et al. Chronic kidney disease, prevalence of premature cardiovascular disease, and relationship to short-term mortality. Am Heart J. 2008 Aug;156(2):277-83.
- Mahon NG, Blackstone EH, Francis GS, Starling RC 3rd, Young JB, Lauer MS. The prognostic value of estimated creatinine clearance alongside functional capacity in ambulatory patients with chronic congestive heart failure. J Am Coll Cardiol. 2002;40:1106–13.
- Muntner P, He J, Hamm L, Loria C, Whelton PK. Renal insufficiency and subsequent death resulting from cardiovascular disease in the United States. J Am Soc Nephrol. 2002;13(3):745-53.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303-10.
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546-54.
- Alberti C, Brun-Buisson C, Burchardi H, Martin C, Goodman S, Artigas A, et al. Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study. Intensive Care Med. 2002;28:108–121.
- Bagshaw SM, Lapinsky S, Dial S, Arabi Y, Dodek P, Wood G, et al. Acute kidney injury in septic shock: clinical outcomes and impact of duration of hypotension prior to initiation of antimicrobial therapy. Intensive Care Med. 2009;35(5):871-81.
- Lopes JA, Jorge S, Resina C, Santos C, Pereira A, Neves J, et al. Acute renal failure in patients with sepsis. Crit Care. 2007;11(2):411.
- Bagshaw SM, George C, Bellomo R. Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care. 2008;12(2):R47.
- Oppert M, Engel C, Brunkhorst FM, Bogatsch H, Reinhart K, Frei U, et al. Acute renal failure in patients with severe sepsis and septic shock--a significant independent risk factor for mortality: results from the German Prevalence Study. Nephrol Dial Transplant. 2008;23(3):904-9.
- Charpentier J, Luyt CE, Fulla Y, Vinsonneau C, Cariou A, Grabar S, et al. Brain natriuretic peptide: A marker of myocardial dysfunction and prognosis during severe sepsis. Crit Care Med. 2004;32(3):660-5.
- Jardin F, Fourme T, Page B, Loubières Y, Vieillard-Baron A, Beauchet A, et al. Persistent preload defect in severe sepsis despite fluid loading: A longitudinal echocardiographic study in patients with septic shock. Chest. 1999;116(5):1354-9.
- ver Elst KM, Spapen HD, Nguyen DN, Garbar C, Huyghens LP, et al. Cardiac troponins I and T are biological markers of left ventricular dysfunction in septic shock. Clin Chem. 2000;46(5):650-7.
- Arlati S, Brenna S, Prencipe L, Marocchi A, Casella GP, Lanzani M, et al. Myocardial necrosis in ICU patients with acute non-cardiac disease: a prospective study. Intensive Care Med. 2000 Jan;26(1):31-7.
- Mehta NJ, Khan IA, Gupta V, Jani K, Gowda RM, Smith PR. Cardiac troponin I predicts myocardial dysfunction and adverse outcome in septic shock. Int J Cardiol. 2004;95(1):13-7.
- Ammann P, Maggiorini M, Bertel O, Haenseler E, Joller-Jemelka HI, Oechslin E, et al. Troponin as a risk factor for mortality in critically ill patients without acute coronary syndromes. J Am Coll Cardiol. 2003 Jun 4;41(11):2004-9.
- Georgopoulou T, Petrakis I, Dermitzaki K, Pleros C, Drosataki E, et al. Cardiorenal Syndrome: Challenges in Everyday Clinical Practice and Key Points towards a Better Management. J Clin Med. 2023;12(12):4121.
- Kim CJ, Choi IJ, Park HJ, Kim TH, Kim PJ, Chang K, et al. Impact of Cardiorenal Anemia Syndrome on Short- and Long-Term Clinical Outcomes in Patients Hospitalized with Heart Failure. Cardiorenal Med. 2016;6(4):269-78.
- Silverberg DS, Wexler D, Blum M, Keren G, Sheps D, Leibovitch E, et al. The use of subcutaneous erythropoietin and intravenous iron for the treatment of the anemia of severe, resistant congestive heart failure improves cardiac and renal function and functional cardiac class, and markedly reduces hospitalizations. J Am Coll Cardiol 2000;35(7)1737-44
- Groenveld HF, Januzzi JL, Damman K, van Wijngaarden J, Hillege HL, van Veldhuisen DJ, et al. Anemia and mortality in heart failure patients a systematic review and meta-analysis. J Am Coll Cardiol. 2008;52(10):818-27.
- Young JB, Abraham WT, Albert NM, Gattis Stough W, Gheorghiade M, Greenberg BH, et al. Relation of Low Hemoglobin and Anemia to Morbidity and Mortality in Patients Hospitalized With Heart Failure (Insight from the OPTIMIZE-HF Registry). The American Journal of Cardiology 2008;101(2):223-30.
- Palazzuoli A, Antonelli G, Nuti R. Anemia in Cardio-Renal Syndrome: clinical impact and pathophysiologic mechanisms. Heart Fail Rev 2011;16(6):603-7.
- Adams KF Jr, Patterson JH, Oren RM, Mehra MR, O'Connor CM, Piña IL, et al. Prospective assessment of the occurrence of anemia in patients with heart failure: Results from the Study of Anemia in a Heart Failure Population (STAMINA-HFP) Registry. American Heart Journal 2009;157(5):926-32.
- van der Putten K, Braam B, Jie KE, Gaillard CA. Mechanisms of Disease: erythropoietin resistance in patients with both heart and kidney failure. Nat Clin Pract Nephrol. 2008;4(1):47-57.
- Belonje AM, Voors AA, van der Meer P, van Gilst WH, Jaarsma T, van Veldhuisen DJ. Endogenous erythropoietin and outcome in heart failure. Circulation. 2010;121(2):245-51.
- Handelman GJ, Levin NW. Iron and anemia in human biology: a review of mechanisms. Heart Fail Rev. 2008;13(4):393-404. doi: 10.1007/s10741-008-9086-x.
- KDOQI; National Kidney Foundation. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Anemia in Chronic Kidney Disease. Am J Kidney Dis. 2006 ;47(5 Suppl 3):S11-145.
- Grune T, Sommerburg O, Siems WG. Oxidative stress in anemia. Clin Nephrol. 2000;53(1):S18-22.
- Brezis M, Rosen S. Hypoxia of the Renal Medulla — Its Implications for Disease. New England Journal of Medicine 1995;332(10):647–655.
- Denton KM, Shweta A, Anderson WP. Preglomerular and Postglomerular Resistance Responses to Different Levels of Sympathetic Activation by Hypoxia. JASN 2002;13(1):27-34.
- Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, et al. Correction of Anemia with Epoetin Alfa in Chronic Kidney Disease. New England Journal of Medicine 2006;355(20):2085–2098.
- Drüeke TB, Locatelli F, Clyne N, Eckardt KU, Macdougall IC, Tsakiris D, Burger HU, et al. Normalization of Hemoglobin Level in Patients with Chronic Kidney Disease and Anemia. New England Journal of Medicine 2006;355(20):2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, et al. A Trial of Darbepoetin Alfa in Type 2 Diabetes and Chronic Kidney Disease. New England Journal of Medicine 2009;361(21):2019-32.
- Swedberg K, Young JB, Anand IS, Cheng S, Desai AS, Diaz R, et al. Treatment of Anemia with Darbepoetin Alfa in Systolic Heart Failure. New England Journal of Medicine 2013;368(13):1210-9.
- McCullough PA. Anemia of cardiorenal syndrome. Kidney Int Suppl (2011). 2021;11(1):35-45.
- House AA, Wanner C, Sarnak MJ, Piña IL, McIntyre CW, Komenda P, et al. Heart failure in chronic kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019 Jun;95(6):1304-1317.
- Ponikowski P, Filippatos G, Colet JC, Willenheimer R, Dickstein K, Lüscher T, et al. The impact of intravenous ferric carboxymaltose on renal function: an analysis of the FAIR-HF study. Eur J Heart Fail. 2015;17(3):329-39.
- Ku E, Del Vecchio L, Eckardt KU, Haase VH, Johansen KL, Nangaku M, et al. Novel anemia therapies in chronic kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2023;104(4):655-680.
- Anker SD, Comin Colet J, Filippatos G, Willenheimer R, Dickstein K, Drexler H,et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med. 2009;361(25):2436-48.
- Ben-Assa E, Shacham Y, Shashar M, Leshem-Rubinow E, Gal-Oz A, Schwartz IF, et al. Target Hemoglobin May Be Achieved with Intravenous Iron Alone in Anemic Patients with Cardiorenal Syndrome: An Observational Study. Cardiorenal Med. 2015;5(4):246-53.
- Swedberg K, Young JB, Anand IS, Cheng S, Desai AS, Diaz R, et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med. 2013 ;368(13):1210-9.
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution . Eur J Hear Fail 2016;18(8)891-975.
- Locatelli F, Del Vecchio L, Esposito C, et al. Consensus commentary and position of the Italian Society of Nephrology on KDIGO controversies conference on novel anemia therapies in chronic kidney disease. J Nephrol. 2024.
- Locatelli F, Bárány P, Covic A, De Francisco A, Del Vecchio L, Goldsmith D, et al. Kidney Disease: Improving Global Outcomes guidelines on anaemia management in chronic kidney disease: a European Renal Best Practice position statement. Nephrol Dial Transplant. 2013;28(6):1346-59.
- Jackevicius CA, Co MJ, Warner AL. Predictors of erythropoietin use in patients with cardiorenal anaemia syndrome. Int J Pharm Pract. 2015;23(3):199-204.
- Kaplan JM, Sharma N, Dikdan S. Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease. Int J Mol Sci. 2018;19(2):389.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9(6):677-84.
- Cicoira M, Bolger AP, Doehner W, Rauchhaus M, Davos C, Sharma R, et al. High tumour necrosis factor-alpha levels are associated with exercise intolerance and neurohormonal activation in chronic heart failure patients. Cytokine. 2001;15:80-6.
- Glance LG, Wissler R, Mukamel DB, Li Y, Diachun CA, Salloum R, et al. Perioperative outcomes among patients with the modified metabolic syndrome who are undergoing noncardiac surgery. Anesthesiology. 2010, 113:859-72.
- Scabbia EV, Scabbia L. The cardio-renal syndrome (CRS). IJC Metab Endoc. 2015;9:1-4. http://creativecommons.org/licenses/by-nc-nd/3.0/. Accessed May 2024.
- Marassi M, Fadini GP. The cardio-renal-metabolic connection: a review of the evidence. Cardiovasc Diabetol. 2023;22(1)195.
- Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pr 2022;183109119.
- Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Fail. 2020;22(8):1342-1356.
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395(10225):709-33.
- Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, et al. Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology. Eur Hear J. 2018;39(48)4243-4254.
- Seferović PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, Bauersachs J, et al. Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Hear Fail 2018;20(5)853-872.
- Usman MS, Khan MS, Butler J. The Interplay Between Diabetes, Cardiovascular Disease, and Kidney Disease. 2021. In: Chronic Kidney Disease and Type 2 Diabetes. Arlington (VA): American Diabetes Association; 2021. Bookshelf ID NBK571718.
- Damman K, Valente MA, Voors AA, O'Connor CM, van Veldhuisen DJ, Hillege HL. et al. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Hear J. 2014;35(7)455-69.
- Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circ 2021;143(11)1157-1172.
- Bock JS, Gottlieb SS. Cardiorenal syndrome: new perspectives. Circulation. 2010;121(23):2592-600.
- Burke M, Pabbidi MR, Farley J, Roman RJ. Molecular mechanisms of renal blood flow autoregulation. Curr Vasc Pharmacol. 2014;12(6):845-58.
- Hanberg JS, Sury K, Wilson FP, Brisco MA, Ahmad T, Ter Maaten JM, et al. Reduced Cardiac Index Is Not the Dominant Driver of Renal Dysfunction in Heart Failure. J Am Coll Cardiol. 2016;67(19):2199-208.
- Cowie MR, Komajda M, Murray-Thomas T, Underwood J, Ticho B. Prevalence and impact of worsening renal function in patients hospitalized with decompensated heart failure: results of the prospective outcomes study in heart failure (POSH).Eur Heart J. 2006;27(10):1216–22.
- Butler J, Forman DE, Abraham WT, Gottlieb SS, Loh E, Massie BM, et al. Relationship between heart failure treatment and development of worsening renal function among hospitalized patients. Am Heart J. 2004;147(2):331-8.
- Weinfeld MS, Chertow GM, Stevenson LW. Aggravated renal dysfunction during intensive therapy for advanced chronic heart failure. Am Heart J. 1999;138(2):285-90.
- Grams ME, Astor BC, Bash LD, Matsushita K, Wang Y, Coresh J. Albuminuria and estimated glomerular filtration rate independently associate with acute kidney injury. J Am Soc Nephrol. 2010;21(10):1757-64.
- Sowers JR, Whaley-Connell A, Hayden MR. The Role of Overweight and Obesity in the Cardiorenal Syndrome. Cardiorenal Med. 2011;1(1):5-12.
- Ronco C, Cicoira M, McCullough PA. Cardiorenal syndrome type 1: pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J Am Coll Cardiol. 2012;60(12):1031-42.
- Hunley TE, Ma LJ, Kon V. Scope and mechanisms of obesity-related renal disease. Curr Opin Nephrol Hypertens. 2010;19(3):227-34.
- Aune D, Sen A, Norat T, Janszky I, Romundstad P, Tonstad S, Vatten LJ. Body Mass Index, Abdominal Fatness, and Heart Failure Incidence and Mortality: A Systematic Review and Dose-Response Meta-Analysis of Prospective Studies. Circulation. 2016;133(7):639-49.
- Uduman J. Epidemiology of Cardiorenal Syndrome. Adv Chronic Kidney Dis. 2018;25(5):391-9.
- Vardeny O, Wu DH, Desai A, Rossignol P, Zannad F, Pitt B, et al. Influence of baseline and worsening renal function on efficacy of spironolactone in patients With severe heart failure: insights from RALES (Randomized Aldactone Evaluation Study). J Am Coll Cardiol. 2012;60(20):2082-9.
- Palazzuoli A, Testani JM, Ruocco G, Pellegrini M, Ronco C, Nuti R. Different diuretic dose and response in acute decompensated heart failure: Clinical characteristics and prognostic significance. Int J Cardiol. 2016;224:213-9. doi: 10.1016/j.ijcard.2016.09.005.
- Ellison DH, Felker GM. Diuretic Treatment in Heart Failure. N Engl J Med. 2017;377(20):1964-1975.
- Hostetter TH, Pfeffer JM, Pfeffer MA, Dworkin LD, Braunwald E, Brenner BM. Cardiorenal hemodynamics and sodium excretion in rats with myocardial infarction. American Journal of Physiology-Heart and Circulatory Physiology 1983;245(1):H98–H103.
- Binanay C, Califf RM, Hasselblad V, O'Connor CM, Shah MR, Sopko G, et al; ESCAPE Investigators and ESCAPE Study Coordinators. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: the ESCAPE trial. JAMA 2005;294(13):1625–1633.
- Tarvasmäki T, Haapio M, Mebazaa A, Sionis A, Silva-Cardoso J, Tolppanen H, et al. Acute kidney injury in cardiogenic shock: definitions, incidence, haemodynamic alterations, and mortality. European Journal of Heart Failure 2018;20(3):572–581.
- Lauschke A, Teichgräber UKM, Frei U, Eckardt K-U. ‘Low-dose’ dopamine worsens renal perfusion in patients with acute renal failure. Kidney International 2006;69(9):1669–1674.
- Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased Central Venous Pressure Is Associated With Impaired Renal Function and Mortality in a Broad Spectrum of Patients With Cardiovascular Disease. Journal of the American College of Cardiology 2009;53(7):582–588.
- Dalfino L, Tullo L, Donadio I, Malcangi V, Brienza N. Intra-abdominal hypertension and acute renal failure in critically ill patients. Intensive Care Med. 2008;34(4):707-13.
- Damman K, Valente MAE, van Veldhuisen DJ, Cleland JGF, O'Connor CM, Metra M,et al. Plasma neutrophil gelatinase-associated lipocalin and predicting clinically relevant worsening renal function in acute heart failure. Int J Mol Sci. 2017; 18(7):1470.
- Burnett JC Jr, Knox FG. Renal interstitial pressure and sodium excretion during renal vein constriction. Am J Physiol. 1980, 238:F279-82.
- Afsar B, Ortiz A, Covic A, Solak Y, Goldsmith D, Kanbay M. Focus on renal congestion in heart failure. Clin Kidney J. 2016;9(1):39-47.
- Verbrugge FH, Dupont M, Steels P, Grieten L, Malbrain M, Tang WH, Mullens W. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J Am Coll Cardiol 2013;62(6):485-95.
- Alpert JS. The effect of right ventricular dysfunction on left ventricular form and function. Chest 2001;119(6)1632-3.
- Konstam, MA, Isner, J. The Right Ventricle, Konstam, MA, Isner, J (Eds), Kluwer Academic Publishers, 2009.
- Marcus JT, Vonk Noordegraaf A, Roeleveld RJ, Postmus PE, Heethaar RM, Van Rossum AC, et al. Impaired left ventricular filling due to right ventricular pressure overload in primary pulmonary hypertension: noninvasive monitoring using MRI. Chest 2001;119(6)1761-5.
- Little WC, Badke FR, O'Rourke RA. Effect of right ventricular pressure on the end-diastolic left ventricular pressure-volume relationship before and after chronic right ventricular pressure overload in dogs without pericardia. Circ Res 1984;54(6)719-30.
- Testani JM, Khera AV, St John Sutton MG, Keane MG, Wiegers SE, Shannon RP, et al. Effect of right ventricular function and venous congestion on cardiorenal interactions during the treatment of decompensated heart failure. Am J Cardiol 2010; ;105(4)511-6.
- Chinnappa S, Tu Y-K, Yeh YC, Glorieux G, Vanholder R, Mooney A. Association between Protein-Bound Uremic Toxins and Asymptomatic Cardiac Dysfunction in Patients with Chronic Kidney Disease. Toxins (Basel) 2018;10(12).
- Lin CJ, Liu HL, Pan CF, Chuang CK, Jayakumar T, Wang TJ, Chen HH, et al. Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch Med Res 2012;43(6):451–456.
- Wu IW, Hsu KH, Lee CC, Sun CY, Hsu HJ, Tsai CJ, et al. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant 2011;26(3):938–947.
- D'Oria R, Schipani R, Leonardini A, Natalicchio A, Perrini S, Cignarelli A, et al. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid Med Cell Longev. 2020;2020:5732956.
- Owada S, Goto S, Bannai K, Hayashi H, Nishijima F, Niwa T. Indoxyl Sulfate Reduces Superoxide Scavenging Activity in the Kidneys of Normal and Uremic Rats. AJN 2008;28(3):446–454.
- Lekawanvijit S, Krum H. Cardiorenal syndrome: acute kidney injury secondary to cardiovascular disease and role of protein-bound uraemic toxins. J Physiol 2014;592(Pt 18):3969–3983.
- D'Oria R, Schipani R, Leonardini A, Natalicchio A, Perrini S, Cignarelli A, et al. Serum Indoxyl Sulfate Is Associated with Vascular Disease and Mortality in Chronic Kidney Disease Patients. CJASN 2009;4(10):1551-8.
- Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011;121(11):4393–4408.
- Reddy SSK, Chaiban JT. THE ENDOCRINOLOGY OF AGING: A KEY TO LONGEVITY “GREAT EXPECTATIONS”. Endocr Pract. 2017;23(9):1107-1116.
- Donovan K, Herrington WG, Paré G, Pigeyre M, Haynes R, Sardell R, et al. Fibroblast Growth Factor-23 and Risk of Cardiovascular Diseases: A Mendelian Randomization Study. Clin J Am Soc Nephrol. 2023;18(1)17-27.
- Kao YH, Chen YC, Lin YK, Shiu RJ, Chao TF, Chen SA, Chen YJ. FGF-23 dysregulates calcium homeostasis and electrophysiological properties in HL-1 atrial cells. Eur J Clin Invest 2014;44(8):795–801.
- Lewiecki EM, Bilezikian JP, Khosla S, Marcus R, McClung MR, Miller PD, et al. Osteoporosis update from the 2010 santa fe bone symposium. J Clin Densitom. 2011;14(1)1-21.
- Borovac JA, D'Amario D, Bozic J, Glavas D. Sympathetic nervous system activation and heart failure: Current state of evidence and the pathophysiology in the light of novel biomarkers. World J Cardiol. 2020;12(8):373-408.
- Floras JS, Ponikowski P. The sympathetic/parasympathetic imbalance in heart failure with reduced ejection fraction. Eur Heart J. 2015;36(30):1974-82b.
- Heesch CM. Reflexes that control cardiovascular function. Am J Physiol. 1999;277(6):234-43.
- Kopp UC. Neural Control of Renin Secretion Rate Morgan & Claypool Life Sciences; 2011. https://www.ncbi.nlm.nih.gov/books/NBK57240/. Accessed April, 2024.
- de Lucia C, Piedepalumbo M, Paolisso G, Koch WJ. Sympathetic nervous system in age-related cardiovascular dysfunction: Pathophysiology and therapeutic perspective. Int J Biochem Cell Biol. 2019;108:29-33.
- Zhang DY, Anderson AS. The sympathetic nervous system and heart failure. Cardiol Clin. 2014;32(1):33-45.
- riposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54:1747-62.
- Triposkiadis F, Karayannis G, Giamouzis G, et al. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54(19):1747-62.
- Feng QP, Hedner T, Andersson B, Lundberg JM, Waagstein F. Cardiac neuropeptide Y and noradrenaline balance in patients with congestive heart failure. Br Heart J. 1994;71(3):261-7.
- Herring N, Cranley J, Lokale MN, Li D, Shanks J, Alston EN, et al. The cardiac sympathetic co-transmitter galanin reduces acetylcholine release and vagal bradycardia: implications for neural control of cardiac excitability. J Mol Cell Cardiol. 2012;52(3):667-76.
- Herring N, Cranley J, Lokale MN, Li D, Shanks J, Alston EN, et al. Contribution of the endothelin system to the renal hypoperfusion associated with experimental congestive heart failure. J Cardiovasc Pharmacol. 1999;34(4):612-7.
- Mahata SK, O'Connor DT, Mahata M, Yoo SH, Taupenot L, Wu H, et al. Novel autocrine feedback control of catecholamine release. A discrete chromogranin a fragment is a noncompetitive nicotinic cholinergic antagonist. J Clin Invest. 1997;100(6):1623-33.
- Ahmad Y, Francis DP, Bhatt DL, Howard JP. Renal Denervation for Hypertension: A Systematic Review and Meta-Analysis of Randomized, Blinded, Placebo-Controlled Trials. JACC Cardiovasc Interv. 2021;14(23):2614-2624.
- Roubsanthisuk W, Kunanon S, Chattranukulchai P, Panchavinnin P, Wongpraparut N, Chaipromprasit J, et al. 2022 Renal denervation therapy for the treatment of hypertension: a statement from the Thai Hypertension Society. Hypertens Res. 2023;46(4)898-912.
- Kim J, Padanilam BJ. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J Am Soc Nephrol. 2013;24(2):229-42.
- Wills LP, Trager RE, Beeson GC, Lindsey CC, Peterson YK, Beeson CC, et al. The β2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J Pharmacol Exp Ther. 2012;342(1):106-18.
- Jesinkey SR, Funk JA, Stallons LJ, Wills LP, Megyesi JK, Beeson CC, et al. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol. 2014;25(6):1157-62.
- Hasegawa S, Inoue T, Nakamura Y, Fukaya D, Uni R, Wu CH, et al. Activation of Sympathetic Signaling in Macrophages Blocks Systemic Inflammation and Protects against Renal Ischemia-Reperfusion Injury. J Am Soc Nephrol. 2021;32(7):1599-1615.
- Doi K, Matsuura R. Sympathetic Nerve Activation in Acute Kidney Injury and Cardiorenal Syndrome. Nephron. 2023;147(12):717-720.
- Damman K, Navis G, Voors AA, Asselbergs FW, Smilde TD, Cleland JG, et al. Worsening renal function and prognosis in heart failure: systematic review and meta-analysis. J Card Fail. 2007;13(8):599-608.
- Jois P, Mebazaa A. Cardio-renal syndrome type 2: epidemiology, pathophysiology, and treatment. Semin Nephrol. 2012;32(1):26-30.
- Matsuura R, Yamashita T, Hayase N, Hamasaki Y, Noiri E, Numata G, et al. Preexisting heart failure with reduced ejection fraction attenuates renal fibrosis after ischemia reperfusion via sympathetic activation. Sci Rep. 2021;11(1):15091.
- Harrison-Bernard LM. The renal renin-angiotensin system. Advances in Physiology Education 2009;33(4):270-4.
- Johnson MD, Malvin RL. Stimulation of renal sodium reabsorption by angiotensin II. Am J Physiol 1977;232(4):298-306.
- Barton M, Shaw S, d'Uscio LV, Moreau P, Lüscher TF.Angiotensin II increases vascular and renal endothelin-1 and functional endothelin converting enzyme activity in vivo: role of ETA receptors for endothelin regulation. Biochem Biophys Res Commun. 1997;238(3):861-5.
- Neuhofer W, Pittrow D. Role of endothelin and endothelin receptor antagonists in renal disease. European Journal of Clinical Investigation 2006;36(s3):78-88.
- Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-β1 and endothelin-1 from fibroblasts. Cardiovascular research 1998;40(2):352-63.
- Hitomi H, Kiyomoto H, Nishiyama A. Angiotensin II and oxidative stress. Curr Opin Cardiol. 2007 Jul;22(4):311-5..
- Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The Sympathetic Nervous System in Heart Failure: Physiology, Pathophysiology, and Clinical Implications. Journal of the American College of Cardiology 2009;54(19):1747-62. doi: 10.1016/j.jacc.2009.05.015.
- Funaya H, Kitakaze M, Node K, Minamino T, Komamura K, Hori M. Plasma Adenosine Levels Increase in Patients With Chronic Heart Failure. Circulation 1997;95(6):1363-5.
- Massie BM, O'Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, et al. Rolofylline, an Adenosine A1−Receptor Antagonist, in Acute Heart Failure. New England Journal of Medicine 2010;363(15):1419-28.
- Torres VE. Vasopressin in chronic kidney disease, an elephant in the room? Kidney Int 2009;76(9):925-28.
- Bardoux P, Martin H, Ahloulay M, Schmitt F, Bouby N, Trinh-Trang-Tan MM, et al. Vasopressin contributes to hyperfiltration, albuminuria, and renal hypertrophy in diabetes mellitus: study in vasopressin-deficient Brattleboro rats. Proc Natl Acad Sci USA 1999;96(18):10397-402.
- Rouleau JL, Packer M, Moyé L, de Champlain J, Bichet D, Klein M, Rouleau JR, et al. Prognostic value of neurohumoral activation in patients with an acute myocardial infarction: effect of captopril. J Am Coll Cardiol. 1994;24(3):583-91.
- Kazory A, Costanzo MR. The dynamic relationship between serum chloride and cardiorenal syndrome. Rev Cardiovasc Med. 2020;21(1):25-29.
- Rinehart J, Kahle KT, de Los Heros P, Vazquez N, Meade P, Wilson FH, et al. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl− cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci USA. 2005;10216777–16782.
- Liang L, Shimosawa T. Molecular Mechanisms of Na-Cl Cotransporter in Relation to Hypertension in Chronic Kidney Disease. Int J Mol Sci. 2022;24(1):286. doi: 10.3390/ijms24010286.
- Berend K, van Hulsteijn LH, Gans RO. Chloride: the queen of electrolytes? Eur J Intern Med. 2012;23(3):203-11.
- Hanberg JS, Rao V, Ter Maaten JM, Laur O, Brisco MA, Perry Wilson F, et al. Hypochloremia and Diuretic Resistance in Heart Failure: Mechanistic Insights. Circ Heart Fail. 2016;9(8):1-12.
- Ter Maaten JM, Damman K, Hanberg JS, Givertz MM, Metra M, O'Connor CM,, et al. Hypochloremia, Diuretic Resistance, and Outcome in Patients With Acute Heart Failure. Circ Heart Fail. 2016;9(8):e003109.
- Kotchen TA, Luke RG, Ott CE, Galla JH, Whitescarver S. Effect of chloride on renin and blood pressure responses to sodium chloride. Ann Intern Med. 1983;98(5 Pt 2):817-22.
- Wilcox CS. Regulation of renal blood flow by plasma chloride. J Clin Invest. 1983;71(3):726-35.
- Cuthbert JJ, Pellicori P, Rigby A, Pan D, Kazmi S, Shah P, et al. Low serum chloride in patients with chronic heart failure: clinical associations and prognostic significance. Eur J Heart Fail. 2018;20(10):1426-1435.
- Grodin JL, Simon J, Hachamovitch R, Wu Y, Jackson G, Halkar M, et al. Prognostic Role of Serum Chloride Levels in Acute Decompensated Heart Failure. J Am Coll Cardiol. 2015;66(6):659-66.
- Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82(2):291-5.
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. American Journal of Physiology-Lung Cellular and Molecular Physiology 2000;279(6):L1005–L1028.
- Grodin JL, Simon J, Hachamovitch R, Wu Y, Jackson G, Halkar M, et al. Venous congestion, endothelial and neurohormonal activation in acute decompensated heart failure: cause or effect? Curr Heart Fail Rep. 2015;12(3):215-22.
- Katz AM, Konstam MA. Heart failure: pathophysiology, molecular biology, and clinical management, 2nd edn. Lippincott Williams & Wilkins, Philadelphia, 2009.
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, et al. Role of NAD(P)H oxidase-and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 2005; 45(5):860-6.
- Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. Journal of molecular and cellular cardiology 2003; 35(7):851-9.
- Becker BN, Himmelfarb J, Henrich WL, Hakim RM. Reassessing the cardiac risk profile in chronic hemodialysis patients: a hypothesis on the role of oxidant stress and other non-traditional cardiac risk factors. J Am Soc Nephrol. 1997;8(3):475-86.
- Kraut EJ, Chen S, Hubbard NE, Erickson KL, Wisner DH. Tumor necrosis factor depresses myocardial contractility in endotoxemic swine. J Trauma. 1999, 46:900-6.
- Radeke HH, Meier B, Topley N, Flöge J, Habermehl GG, Resch K. Interleukin 1-alpha and tumor necrosis factor-alpha induce oxygen radical production in mesangial cells. Kidney Int. 1990;37(2):767-75.
- Wettersten N, Maisel AS. Biomarkers for Heart Failure: An Update for Practitioners of Internal Medicine. Am J Med 2016;129(6):560-7.
- Barreto DV, Barreto FC, Liabeuf S, Temmar M, Lemke HD, Tribouilloy C, et al. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney International 2010;77(6):550-6.
- Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, et al. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int 2005;67(4):1216–1233.
- Pereira BJ, Shapiro L, King AJ, Falagas ME, Strom JA, Dinarello CA. Plasma levels of IL-1 beta, TNF alpha and their specific inhibitors in undialyzed chronic renal failure, CAPD and hemodialysis patients. Kidney Int. 1994;45(3):890-6.
- Arici M, Walls J. End-stage renal disease, atherosclerosis, and cardiovascular mortality: Is C-reactive protein the missing link? Kidney International 2001;59(2):407–14.
- Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C-reactive protein induces human peripheral blood monocytes to synthesize tissue factor. Blood. 1993;82(2):513-20.
- Minami Y, Kajimoto K, Sato N, Hagiwara N; ATTEND Study Investigators. Effect of Elevated C-Reactive Protein Level at Discharge on Long-Term Outcome in Patients Hospitalized for Acute Heart Failure. The American Journal of Cardiology 2018;121(8):961-8.
- Kim BS, Jeon DS, Shin MJ, Kim YO, Song HC, Lee SH, et al. Persistent Elevation of C-Reactive Protein May Predict Cardiac Hypertrophy and Dysfunction in Patients Maintained on Hemodialysis. AJN 2005;25(3):189-95.
- Di Lullo L, Bellasi A, Barbera V, Russo D, Russo L, Di Iorio B, et al. Pathophysiology of the cardio-renal syndromes types 1–5: An uptodate. Indian Heart Journal 2017;69(2):255-65.
- Wang CS, FitzGerald JM, Schulzer M, et al. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA. 2005;294(15):1944-56.
- Gehlbach BK, Geppert E. The pulmonary manifestations of left heart failure. Chest. 2004;125(2):669-82.
- Schanz M, Shi J, Wasser C, Alscher MD, Kimmel M. Urinary [TIMP-2] × [IGFBP7] for risk prediction of acute kidney injury in decompensated heart failure. Clin Cardiol. 2017;40(7):485-91.
- Ahmed MM, Tazyeen S, Alam A, Farooqui A, Ali R, Imam N, et al. Deciphering key genes in cardio-renal syndrome using network analysis. Bioinformation. 2021;17(1):86-100.
- Maisel AS, Katz N, Hillege HL, Shaw A, Zanco P, Bellomo R, et al. Biomarkers in kidney and heart disease. Nephrol Dial Transplant. 2011;26(1):62-74.
- Oliveros E, Oni ET, Shahzad A, Kluger AY, Lo KB, Rangaswami J, McCullough PA. Benefits and Risks of Continuing Angiotensin-Converting Enzyme Inhibitors, Angiotensin II Receptor Antagonists, and Mineralocorticoid Receptor Antagonists during Hospitalizations for Acute Heart Failure. Cardiorenal Med. 2020;10(2):69-84.
- Bouquegneau A, Krzesinski JM, Delanaye P, Cavalier E. Biomarkers and physiopathology in the cardiorenal syndrome. Clin Chim Acta. 2015;443:100-7.
- Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci. 2011;1243:88-102.
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128.
- Perazella MA, Coca SG, Hall IE, Iyanam U, Koraishy M, Parikh CR. Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients.Clin J Am Soc Nephrol. 2010;5:402–408.
- Haase M, Bellomo R, Devarajan P, Schlattmann P, Haase-Fielitz A; NGAL Meta-analysis Investigator Group. Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis.Am J Kidney Dis. 2009; 54:1012-24.
- Mortara A, Bonadies M, Mazzetti S, Fracchioni I, Delfino P, Chioffi M, et al. Neutrophil gelatinase-associated lipocalin predicts worsening of renal function in acute heart failure: methodological and clinical issues.J Cardiovasc Med (Hagerstown). 2013; 14:629–634.
- Kashani K, Al-Khafaji A, Ardiles T, Artigas A, Bagshaw SM, Bell M, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury.Crit Care. 2013; 17:R25.
- Writing Committee Members; ACC/AHA Joint Committee Members. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure. J Card Fail. 2022;28(5):e1-e167.
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2017;136(6):e137-e161.
- McCullough PA, Duc P, Omland T, McCord J, Nowak RM, Hollander JE, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly Multinational Study.Am J Kidney Dis. 2003; 41:571–579.
- McCullough PA, Neyou A. Comprehensive review of the relative clinical utility of B-type natriuretic peptide and N-terminal pro-B-type natriuretic peptide assays in cardiovascular disease.Open Heart Fail J. 2009; 3:6–17.
- Palazzuoli A, Ruocco G, Pellegrini M, Martini S, Del Castillo G, Beltrami M, et al. Patients with cardiorenal syndrome revealed increased neurohormonal activity, tubular and myocardial damage compared to heart failure patients with preserved renal function.Cardiorenal Med. 2014; 4:257-68.
- Maisel AS, Daniels LB, Anand IS, McCullough PA, Chow SL. Utility of natriuretic peptides to assess and manage patients with heart failure receiving angiotensin receptor blocker/neprilysin inhibitor therapy.Postgrad Med. 2018; 130:299–307.
- Lok DJ, Van Der Meer P, de la Porte PW, Lipsic E, Van Wijngaarden J, Hillege HL, et al. Prognostic value of galectin-3, a novel marker of fibrosis, in patients with chronic heart failure: data from the DEAL-HF study.Clin Res Cardiol. 2010; 99:323–328.
- McCullough PA, Olobatoke A, Vanhecke TE. Galectin-3: a novel blood test for the evaluation and management of patients with heart failure.Rev Cardiovasc Med. 2011; 12:200–210.
- van der Velde AR, Gullestad L, Ueland T, et al. Prognostic value of changes in galectin-3 levels over time in patients with heart failure: data from CORONA and COACH.Circ Heart Fail. 2013; 6:219–226.
- Lee DS, Straus SE, Farkouh ME, Austin PC, Taljaard M, Chong A, et al. Trial of an Intervention to Improve Acute Heart Failure Outcomes. N Engl J Med. 2023;388(1):22-32.
- Colbert G, Jain N, de Lemos JA, Hedayati SS. Utility of traditional circulating and imaging-based cardiac biomarkers in patients with predialysis CKD.Clin J Am Soc Nephrol. 2015; 10:515–529.
- Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, Hillege HL. Drawbacks and prognostic value of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction.Circulation. 2006; 114:1572-80.
- Beigel R, Cercek B, Siegel RJ, Hamilton MA. Echo-Doppler hemodynamics: an important management tool for today’s heart failure care.Circulation. 2015; 131:1031-4.
- Cowie B, Kluger R, Rex S, Missant C. Noninvasive estimation of left atrial pressure with transesophageal echocardiography.Ann Card Anaesth. 2015; 18:312–316.
- Mavrakanas TA, Khattak A, Singh K, Charytan DM. Epidemiology and natural history of the cardiorenal syndromes in a cohort with echocardiography.Clin J Am Soc Nephrol. 2017; 12:1624–1633.
- Kramann R, Erpenbeck J, Schneider RK, Röhl AB, Hein M, Brandenburg VM, et al. Speckle tracking echocardiography detects uremic cardiomyopathy early and predicts cardiovascular mortality in ESRD.J Am Soc Nephrol. 2014; 25:2351-65.
- Hassanin N, Alkemary A. Early detection of subclinical uremic cardiomyopathy using two-dimensional speckle tracking echocardiography.Echocardiography. 2016; 33:527-36.
- Krishnasamy R, Isbel NM, Hawley CM, et al. The association between left ventricular global longitudinal strain, renal impairment and all-cause mortality.Nephrol Dial Transplant. 2014; 29:1218-25.
- Iida N, Seo Y, Sai S, Machino-Ohtsuka T, Yamamoto M, Ishizu T, et al. Clinical implications of intrarenal hemodynamic evaluation by Doppler ultrasonography in heart failure.JACC Heart Fail. 2016; 4:674-82.
- Nijst P, Martens P, Dupont M, Tang WHW, Mullens W. Intrarenal flow alterations during transition from euvolemia to intravascular volume expansion in heart failure patients.JACC Heart Fail. 2017; 5:672-81.
- Faubel S, Patel NU, Lockhart ME, Cadnapaphornchai MA. Renal relevant radiology: use of ultrasonography in patients with AKI.Clin J Am Soc Nephrol. 2014; 9:382-94.
- Edwards NC, Moody WE, Chue CD, Ferro CJ, Townend JN, Steeds RP. Defining the natural history of uremic cardiomyopathy in chronic kidney disease: the role of cardiovascular magnetic resonance.JACC Cardiovasc Imaging. 2014;7:703–714.
- Rutherford E, Talle MA, Mangion K, Bell E, Rauhalammi SM, Roditi G, et al. Defining myocardial tissue abnormalities in end-stage renal failure with cardiac magnetic resonance imaging using native T1 mapping.Kidney Int. 2016; 90:845-52.
- Graham-Brown MP, March DS, Churchward DR, Stensel DJ, Singh A, Arnold R, et al. Novel cardiac nuclear magnetic resonance method for noninvasive assessment of myocardial fibrosis in hemodialysis patients.Kidney Int. 2016; 90:835–844.
- Piccoli A, Codognotto M, Cianci V, Vettore G, Zaninotto M, Plebani M, et al. Differentiation of cardiac and noncardiac dyspnea using bioelectrical impedance vector analysis (BIVA).J Card Fail. 2012; 18:226–232.
- Santarelli S, Russo V, Lalle I, De Berardinis B, Navarin S, Magrini L, et al. Usefulness of combining admission brain natriuretic peptide (BNP) plus hospital discharge bioelectrical impedance vector analysis (BIVA) in predicting 90 days cardiovascular mortality in patients with acute heart faiure. Intern Emerg Med. 2017;12(4):445-451.
- Gnanaraj J, Radhakrishnan J. Cardio-renal syndrome. F1000Res. 2016, 5:
- Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011, 364:797-805.
- Kirklin JK, Naftel DC, Kormos RL, Pagani FD, Myers SL, Stevenson LW, et al. Quantifying the effect of cardiorenal syndrome on mortality after left ventricular assist device implant. J Heart Lung Transplant. 2013;32(12):1205-13.
- Testani JM, Chen J, McCauley BD, Kimmel SE, Shannon RP. Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation. 2010;122(3):265-72.
- Bowman BN, Nawarskas JJ, Anderson JR. Treating Diuretic Resistance: An Overview. Cardiol Rev. 2016;24(5):256-60.
- Uwai Y, Saito H, Hashimoto Y, Inui KI. Interaction and transport of thiazide diuretics, loop diuretics, and acetazolamide via rat renal organic anion transporter rOAT1. J Pharmacol Exp Ther 2000; 295:261–265.
- Hacker K, Maas R, Kornhuber J, Fromm MF, Zolk O. Substrate-Dependent Inhibition of the Human Organic Cation Transporter OCT2: A Comparison of Metformin with Experimental Substrates. PLoS One. 2015;10(9):e0136451.
- Schophuizen CM, Wilmer MJ, Jansen J, Gustavsson L, Hilgendorf C, Hoenderop JG, et al. Cationic uremic toxins affect human renal proximal tubule cell functioning through interaction with the organic cation transporter. Pflugers Arch. 2013;465(12):1701-14.
- Brater DC. Diuretic therapy. N Engl J Med. 1998;339(6):387-95.
- Grodin JL, Stevens SR, de las Fuentes L, Kiernan M, Birati EY, Gupta D, et al. Intensification of Medication Therapy for Cardiorenal Syndrome in Acute Decompensated Heart Failure. J Card Fail. 2016;22(1):26-32.
- Thomson MR, Nappi JM, Dunn SP, Hollis IB, Rodgers JE, Van Bakel AB. Continuous versus intermittent infusion of furosemide in acute decompensated heart failure. J Card Fail. 2010;16(3):188-93.
- Mullens W, Dauw J, Martens P, Verbrugge FH, Nijst P, Meekers E, et al. Acetazolamide in Acute Decompensated Heart Failure with Volume Overload. N Engl J Med. 2022;387(13):1185-95.
- Martens P, Verbrugge FH, Dauw J, Nijst P, Meekers E, Augusto SN, et al. Pre-treatment bicarbonate levels and decongestion by acetazolamide: the ADVOR trial. Eur Heart J. 2023;44(22):1995-2005.
- Cooper LB, Mentz RJ, Gallup D, Lala A, DeVore AD, Vader JM, AbouEzzeddine OF, et al. Serum Bicarbonate in Acute Heart Failure: Relationship to Treatment Strategies and Clinical Outcomes. J Card Fail. 2016;22(9):738-4.
- Martens P, Dauw J, Verbrugge FH, Nijst P, Meekers E, Augusto SN Jr, et al. Decongestion With Acetazolamide in Acute Decompensated Heart Failure Across the Spectrum of Left Ventricular Ejection Fraction: A Prespecified Analysis From the ADVOR Trial. Circulation. 2023;147(3):201-211.
- Meekers E, Dauw J, Martens P, Dhont S, Verbrugge FH, Nijst P, et al. Renal function and decongestion with acetazolamide in acute decompensated heart failure: the ADVOR trial. Eur Heart J. 2023;44(37):3672-82.
- Dhont S, Martens P, Meekers E, Dauw J, Verbrugge FH, Nijst P, et al. Sodium and potassium changes during decongestion with acetazolamide - a pre-specified analysis from the ADVOR trial. Eur J Heart Fail. 2023; ;25(8):1310-9.
- Ng TM, Konopka E, Hyderi AF, Hshieh S, Tsuji Y, Kim BJ, et al. Comparison of bumetanide- and metolazone-based diuretic regimens to furosemide in acute heart failure. J Cardiovasc Pharmacol Ther. 2013;18(4):345-53.
- Jentzer JC, Chawla LS. A Clinical Approach to the Acute Cardiorenal Syndrome. Crit Care Clin. 2015;31(4):685-703.
- Sica DA. Metolazone and its role in edema management. Congest Heart Fail. 2003;9(2):100-5.
- Moranville MP, Choi S, Hogg J, Anderson AS, Rich JD. Comparison of metolazone versus chlorothiazide in acute decompensated heart failure with diuretic resistance. Cardiovasc Ther. 2015;33(2):42-9.
- Steuber TD, Janzen KM, Howard ML. A Systematic Review and Meta-Analysis of Metolazone Compared to Chlorothiazide for Treatment of Acute Decompensated Heart Failure. Pharmacotherapy. 2020;40(9):924-35.
- Verbrugge FH, Dupont M, Bertrand PB, Nijst P, Penders J, Dens J, Verhaert D, et al. Determinants and impact of the natriuretic response to diuretic therapy in heart failure with reduced ejection fraction and volume overload. Acta Cardiol. 2015;70(3):265-73.
- Verbrugge FH, Martens P, Ameloot K, et al. Acetazolamide to increase natriuresis in congestive heart failure at high risk for diuretic resistance. Eur J Heart Fail. 2019 Nov;21(11):1415-1422.
- Martens P, Dauw J, Verbrugge FH, Nijst P, Meekers E, Augusto SN Jr, Ter Maaten JM, et al. Decongestion With Acetazolamide in Acute Decompensated Heart Failure Across the Spectrum of Left Ventricular Ejection Fraction: A Prespecified Analysis From the ADVOR Trial. Circulation. 2023;147(3):201-211.
- Butler J, Anstrom KJ, Felker GM, Givertz MM, Kalogeropoulos AP, Konstam MA, et al. Efficacy and safety of spironolactone in acute heart failure: the ATHENA-HF randomized clinical trial. JAMA Cardiol. 2017;2(9):950-8.
- Kabach M, Alkhawam H, Shah S, Joseph G, Donath EM, Moss N, et al. Ultrafiltration versus intravenous loop diuretics in patients with acute decompensated heart failure: a meta-analysis of clinical trials. Acta Cardiol. 2017;72(2):132-41.
- Pourafshar N, Karimi A, Kazory A. Extracorporeal ultrafiltration therapy for acute decompensated heart failure. Expert Rev Cardiovasc Ther. 2016;14(1):5-13.
- Grodin JL, Carter S, Bart BA, Goldsmith SR, Drazner MH, Tang WH. Direct comparison of ultrafiltration to pharmacological decongestion in heart failure: a per-protocol analysis of CARRESS-HF. Eur J Heart Fail. 2018;20(7):1148-1156.
- Bart BA, Goldsmith SR, Lee KL, Givertz MM, O'Connor CM, Bull DA, et al. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med. 2012;367(24):2296-304.
- Vinod P, Krishnappa V, Chauvin AM, Khare A, Raina R. Heart failure and acute renal dysfunction in the cardiorenal syndrome. Clin Med (Lond). 2020;20(2):146-150.
- Kazory A. Cardiorenal syndrome: ultrafiltration therapy for heart failure--trials and tribulations. Clin J Am Soc Nephrol. 2013;8(10):1816-28.
- Ronco C, Giomarelli P. Current and future role of ultrafiltration in CRS. Heart Fail Rev. 2011;16(6):595-602.
- Chuasuwan A, Kellum JA. Cardio-renal syndrome type 3: epidemiology, pathophysiology, and treatment. Semin Nephrol. 2012;32(1):31-9.
- Vinod P, Krishnappa V, Chauvin AM, Khare A, Raina R. Cardiorenal Syndrome: Role of Arginine Vasopressin and Vaptans in Heart Failure. Cardiol Res. 2017;8(3):87-95.
- Verma D, Firoz A, Garlapati SKP, Sai Charaan Reddy Sathi T, Haris M, et al. Emerging Treatments of Cardiorenal Syndrome: An Update on Pathophysiology and Management. Cureus. 2021;13(8):e17240.
- Obi Y, Kim T, Kovesdy CP, Amin AN, Kalantar-Zadeh K. Current and Potential Therapeutic Strategies for Hemodynamic Cardiorenal Syndrome. Cardiorenal Med. 2016;6(2):83-98.
- Konstam MA, Gheorghiade M, Burnett JC Jr, Grinfeld L, Maggioni AP, Swedberg K, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-31.
- Aghel A, Tang WH. Tolvaptan: the evidence for its therapeutic value in acute heart failure syndrome. Core Evid. 2008;3(1):31-43.
- Bielecka-Dabrowa A, Godoy B, Schefold JC, Koziolek M, Banach M, von Haehling S. Decompensated Heart Failure and Renal Failure: What Is the Current Evidence? Curr Heart Fail Rep. 2018;15(4):224-238.
- Takahama H, Kitakaze M. Pathophysiology of cardiorenal syndrome in patients with heart failure: potential therapeutic targets. Am J Physiol Heart Circ Physiol. 2017;313(4):H715-H721.
- Testani JM, Coca SG, McCauley BD, Shannon RP, Kimmel SE. Impact of changes in blood pressure during the treatment of acute decompensated heart failure on renal and clinical outcomes. Eur J Heart Fail. 2011;13(8):877-84.
- Takeuchi M, Nagai M, Dote K, Kato M, Oda N, Kunita E, et al. Early drop in systolic blood pressure, heart rate at admission, and their effects on worsening renal function in elderly patients with acute heart failure. BMC Cardiovasc Disord. 2020;20(1):366.
- Felker GM, Benza RL, Chandler AB, Leimberger JD, Cuffe MS, et al. Heart failure etiology and response to milrinone in decompensated heart failure: results from the OPTIME-CHF study. J Am Coll Cardiol. 2003;41(6):997-1003.
- Mody BP, Khan MH, Zaid S, Ahn C, Lloji A, Aronow WS, et al. Survival With Continuous Outpatient Intravenous Inotrope Therapy in the Modern Era. Am J Ther. 2020;28(6):e621-e630.
- Yilmaz MB, Yalta K, Yontar C, Karadas F, Erdem A, Turgut OO, Yilmaz A, et al. Levosimendan improves renal function in patients with acute decompensated heart failure: comparison with dobutamine. Cardiovasc Drugs Ther. 2007;21(6):431-5.
- Mebazaa A, Nieminen MS, Packer M, Cohen-Solal A, Kleber FX, Pocock SJ, et al. Levosimendan vs dobutamine for patients with acute decompensated heart failure: the SURVIVE Randomized Trial. JAMA. 2007;297(17):1883-91.
- Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341(10):709-17.
- Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med. 2019;380(4):347-357.
- Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016;375(19)1834-1844.
- Zelniker TA, Braunwald E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J Am Coll Cardiol. 2020;75(4):422-34.
- Herrington WG, Staplin N, Wanner C, Green JB, Hauske SJ, Emberson JR, et al. Empagliflozin in patients with chronic kidney disease. N Engl J Med. 2023;388(2):117–27.
- Solomon SD, McMurray JJV, Claggett B, et al. Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N Engl J Med. 2022;387(12):1089–98.
- Adamson C, Docherty KF, Heerspink HJL, de Boer RA, Damman K, Inzucchi SE, Køber L, et al. Initial decline (dip) in estimated glomerular filtration rate after initiation of dapagliflozin in patients with heart failure and reduced ejection fraction: insights from DAPA-HF. Circulation. 2022; 146(6):438-449.
- Mayne KJ, Staplin N, Keane DF, Wanner C, Brenner S, Cejka V, et al. Effects of empagliflozin on fluid overload, weight, and blood pressure in CKD. J Am Soc Nephrol. 2024;35(2):202–15
- Yeoh SE, Osmanska J, Petrie MC, Brooksbank KJM, Clark AL, Docherty KF, et al. Dapagliflozin vs. metolazone in heart failure resistant to loop diuretics. Eur Heart J. 2023;44(31):2966–77.
- Refardt J, Imber C, Nobbenhuis R, Sailer CO, Haslbauer A, Monnerat S, et al. Treatment Effect of the SGLT2 Inhibitor Empagliflozin on Chronic Syndrome of Inappropriate Antidiuresis: Results of a Randomized, Double-Blind, Placebo-Controlled, Crossover Trial. J Am Soc Nephrol. 2023;34(2):322-332.
- Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373(22):2117-28.
- Neal B, Perkovic V, Matthews DR. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med. 2017;377(21):2099.
- Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med. 2019 J;380(24):2295-2306.
- Kluger AY, Tecson KM, Lee AY, Lerma EV, Rangaswami J, Lepor NE, et al. Class effects of SGLT2 inhibitors on cardiorenal outcomes. Cardiovasc Diabetol. 2019;18(1):99.
- Kalra S, Aydin H, Sahay M, Ghosh S, Ruder S, Tiwaskar M, et al. Cardiorenal Syndrome in Type 2 Diabetes Mellitus - Rational Use of Sodium-glucose Cotransporter-2 Inhibitors. Eur Endocrinol. 2020;16(2):113-121.
- Bailey CJ. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes Metab. 2019;21(6):1291-1298.
- Zelniker TA, Braunwald E. Cardiac and Renal Effects of Sodium-Glucose Co-Transporter 2 Inhibitors in Diabetes: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72(15):1845-55.
- Silva Dos Santos D, Polidoro JZ, Borges-Júnior FA, Girardi AC. Cardioprotection conferred by sodium-glucose cotransporter 2 inhibitors: a renal proximal tubule perspective. Am J Physiol Cell Physiol. 2020 Feb 1;318(2):C328-C36.
- Ussher JR, Drucker DJ. Glucagon-like peptide 1 receptor agonists: cardiovascular benefits and mechanisms of action. Nat Rev Cardiol. 2023;20(7)463-474.
- Alicic RZ, Cox EJ, Neumiller JJ, Tuttle KR. Incretin drugs in diabetic kidney disease: biological mechanisms and clinical evidence. Nat Rev Nephrol. 2021 17(4)227-44.
- Nagahisa T, Saisho Y. Cardiorenal Protection: Potential of SGLT2 Inhibitors and GLP-1 Receptor Agonists in the Treatment of Type 2 Diabetes. Diabetes Ther. 2019;10(5):1733-1752.
- Drucker DJ. Glucagon-like peptide 1 receptor agonists: cardiovascular benefits and mechanisms of action. Nat Rev Cardiol. 2023 ;20(7):463-474.
- Fisman EZ, Tenenbaum A. The dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist tirzepatide: a novel cardiometabolic therapeutic prospect. Cardiovasc Diabetol. 2021;20(1):225.
- Frías JP, Davies MJ, Rosenstock J, et al.Frías JP, Davies MJ, Rosenstock J, et al. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N Engl J Med.2021;385(6):503-515.
- Davis MK, Virani SA. Cardiac resynchronization therapy in the cardiorenal syndrome. Int J Nephrol. 2011;2011:168461.
- Singal G, Upadhyay GA, Borgquist R, Friedman DJ, Chatterjee NA, Kandala J, et al. Renal Response in Patients with Chronic Kidney Disease Predicts Outcome Following Cardiac Resynchronization Therapy. Pacing Clin Electrophysiol. 2015;38(10):1192-200.
- Garg N, Thomas G, Jackson G, Rickard J, Nally JV Jr, Tang WH, et al. Cardiac resynchronization therapy in CKD: a systematic review. Clin J Am Soc Nephrol. 2013;8(8):1293-303.
- Lin G, Gersh BJ, Greene EL, Redfield MM, Hayes DL, Brady PA. Renal function and mortality following cardiac resynchronization therapy. Eur Heart J. 2011;32(2):184-90.
- Gallinoro E, Vanderheyden M, Bartunek J. Device-based therapy of acute cardiorenal syndrome in heart failure. Card Interv Today. 2021;15(3):38–43.
- Rosenblum H, Kapur NK, Abraham WT, Udelson J, Itkin M, Uriel N, et al. Conceptual Considerations for Device-Based Therapy in Acute Decompensated Heart Failure: DRI2P2S. Circ Heart Fail. 2020;13(4):e006731.
- Nathan S, Basir M. Emerging Device Therapies for Cardiorenal Syndrome. JSCAI. 2023;2;101210.
- Beldhuis IE, Streng KW, van der Meer P, Ter Maaten JM, O'Connor CM, Metra M, et al. Trajectories of Changes in Renal Function in Patients with Acute Heart Failure. J Card Fail. 2019 ;25(11):866-874.
- de Silva R, Nikitin NP, Witte KK, Rigby AS, Goode K, Bhandari S, Clark AL, et al. Incidence of renal dysfunction over 6 months in patients with chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J. 2006;27(5):569-81.
- Testani JM, McCauley BD, Chen J, Coca SG, Cappola TP, Kimmel SE. Clinical characteristics and outcomes of patients with improvement in renal function during the treatment of decompensated heart failure. J Card Fail. 2011 ;17(12):993-1000.
- Logeart D, Tabet JY, Hittinger L, Thabut G, Jourdain P, Maison P, et al. Transient worsening of renal function during hospitalization for acute heart failure alters outcome. Int J Cardiol. 2008;127(2):228-32.
- George LK, Koshy SKG, Molnar MZ, Thomas F, Lu JL, Kalantar-Zadeh K, et al. Heart Failure Increases the Risk of Adverse Renal Outcomes in Patients With Normal Kidney Function. Circ Heart Fail 2017; 10(8):e003825.
- Testani JM, McCauley BD, Kimmel SE, Shannon RP. Characteristics of patients with improvement or worsening in renal function during treatment of acute decompensated heart failure. Am J Cardiol. 2010;106(12):1763-9.